关于乙酰氯的国家标准在网上找了很久,找到了份氯乙酰氯的然后发现了乙酰氯GOST 5829-1971乙酰氯 技术条件 (俄罗斯标准)GJB 702-1989 氯乙酰氯 (中国国家军用标准)哪位老师有做过,或者了解乙酰氯国家标准的可否帮下忙,传一份给我,急用,先谢谢大家了
求助 请问哪位大虾有标准品乙酰水杨酸及水杨酸的紫外,非常感谢
问:请问乙酰氯的GC分析方法极其标准?我们是是化工企业,生产高纯乙酰氯,分析量很大,我们目前用GC-FID毛细管柱子,没有标准品,含量大约99。5%,请问这样分析的缺陷,用HPLC行不行?假如有其他微量杂质,是否还要用质谱仪?
关于乙酰氯的国家标准在网上找了很久,找到了份氯乙酰氯的然后发现了乙酰氯GOST 5829-1971乙酰氯 技术条件 (俄罗斯标准)GJB 702-1989 氯乙酰氯 (中国国家军用标准)哪位老师有做过,或者了解乙酰氯国家标准的可否帮下忙,传一份给我,急用,先谢谢大家了
邻氯乙酰乙酰苯胺的国标或行标
食品中甲胺磷和乙酰甲胺磷农药残留量的测定方法1.适用范围本方法适用于谷物、蔬菜和植物油中甲胺磷和乙酰甲胺磷的残留量分析,其最小检出限分别为7.79×10-12g和1.79×10-11g。2.原理概要含有机磷的样品在富氢焰上燃烧,以HPO碎片的形式,放射出波长526nm的特征光,这种特征光通过滤光片选择后,由光电倍增管接收,转换成电信号,经微电流放大器放大后,被记录下来,样品的峰高与标准品的峰高相比,计算出样品相当的含量。3.主要试剂和仪器3.1.主要试剂丙酮;二氯甲烷:重蒸;无水硫酸钠;活性炭:用3mol/L盐酸浸泡过夜,抽滤,用水洗至中性,在120℃下烘干备用;甲胺磷(methamidophos):≥99%;乙酰甲胺磷(acephate):≥99%;甲胺磷和乙酰甲胺磷标准溶液的配制:分别准确称取甲胺磷和乙酰甲胺磷的标准品,用丙酮分别制成0.1mg/mL的标准储备液。使用时用丙酮稀释配制成单一品种的标准使用液(1mg/mL)和混合标准工作液(每个品种浓度为1mg/mL)。贮藏于冰箱中。3.2.仪器气相色谱仪:具有火焰光度检测器;电动振荡器;K-D浓缩器或旋转蒸发器;离心机。4.试样的制备取谷物实验样品经粉碎机粉碎,过20目筛后,制成谷物试样。取蔬菜实验样品洗净,晾干,去掉非食部分后剁碎或经组织捣碎机捣碎,制成蔬菜试样。5.过程简述5.1.提取和净化蔬菜:称取蔬菜试样10g,精确至0.001g,用无水硫酸钠(因蔬菜含水量不同而加入量不同,约50~80g)研磨呈干粉状,倒入具塞锥形瓶中,加入0.2~0.4g活性炭(根据蔬菜色素含量)及80mL丙酮,振摇0.5h,抽滤,滤液浓缩定容至5mL,待气相色谱分析。谷物:称取谷物试样10g,精确至0.001g,置于具塞锥形瓶中,加入40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。小麦:称取小麦试样10g,精确至0.001g,置于具塞锥形瓶中,加入0.2g活性炭及40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。植物油:称取植物油试样5g,用45mL丙酮分次洗入50mL的离心管内,加入5mL水,混匀,在3 000r/min下离心5min,吸取上清液,下面油层再加10mL水和10mL丙酮,离心5min,吸取上清液,合并两次上清液,用K-D浓缩器浓缩近干,残渣和水加入40g无水硫酸钠,研磨呈干粉状,倒入具塞锥形瓶中,加入0.3g活性炭、60mL二氯甲烷,振荡0.5h,抽滤,定容至5mL,待气相色谱分析。5.2.色谱条件色谱柱:玻璃柱,内径3mm,长0.5m,内装2%dEGS/Chromosorb W AWdMCS,80~100mesh。气流:载气,氮气70mL/min,空气0.7kg/cm2,氢气1.2kg/cm2。温度:进样口200℃,柱温180℃。5.3.测定定性:以甲胺磷和乙酰甲胺磷农药标样的保留时间定性。定量:用外标法定量,以甲胺磷和乙酰甲胺磷农药已知浓度的标准样品溶液作外标物,按峰高定量。6.结果计算Xi=hi•Esi•V1hsi•V2•m式中:Xi——样品中i组分有机磷含量,mg/kg;Esi——注入标样中i组分有机磷的含量,ng;hi——样品的峰高,mm;hsi——标样中i组分的峰高,mm;V1——浓缩定容体积,mL;V2——注入色谱样品的体积,μL;m——样品的质量,g。7.方法的精密度添加回收试验中甲胺磷和乙酰甲胺磷的变异系数分别为2.36%和3.95%。8.甲胺磷和乙酰甲胺磷的保留时间在5.2的气相色谱条件下,甲胺磷的保留时间为0.9min,乙酰甲胺磷的保留时间为1.9min。9.来源:GB 14876—94
[font=宋体][size=14px][/size][/font][font='微软雅黑','sans-serif']你好,想问一下老师[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]跑乙酰甲胺磷单标的时候不出峰,跑有机磷混标的时候乙酰甲胺磷可以出峰,换新的一支标准品也是同样的情况,想请教一下老师是哪里有问题,谢谢[/font][font='微软雅黑','sans-serif']求助问题来自微信群。[/font][font=宋体][size=14px][/size][/font]
请问乙酰氯的GC分析方法极其标准?
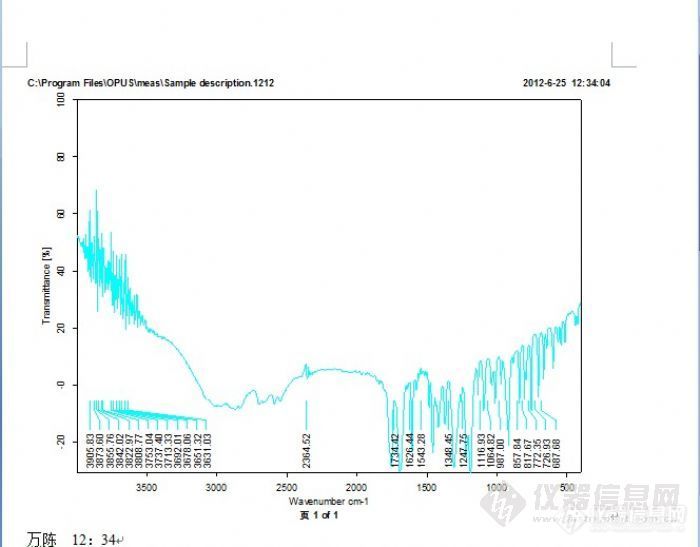
刚做完乙酰水杨酸红外吸收光谱测绘的实验,但之前没学过读图谱,马上要交报告了,望个高手帮帮忙啊!非常谢谢!我测的谱图在附件上。问题:1.根据乙酰水杨酸红外谱图进行图谱分析,分别指出3300~2500 cm-1处的宽带、1754 cm-1、1691 cm-1、1606cm-1、1574 cm-1、1485 cm-1、1308 cm-1、1189 cm-1以及 756 cm-1等吸收峰的归属。 2.标准的乙酰水杨酸红外谱图对照,指出相似度有多大。http://ng1.17img.cn/bbsfiles/images/2012/07/201207011129_375245_2511548_3.jpg
1.根据乙酰水杨酸红外谱图进行图谱分析,分别指出3300~2500 cm-1处的宽带、1754 cm-1、1691 cm-1、1606cm-1、1574 cm-1、1485 cm-1、1308 cm-1、1189 cm-1以及 756 cm-1等吸收峰的归属。 2.标准的乙酰水杨酸红外谱图对照,指出相似度有多大。
现我厂采购两种乙酰氯,一种是有磷的,一种是无磷的,用酯化法检测后有磷乙酰氯与生产厂家结果分析误差比较大,什么原因?各位谁知道
如题:乙酰甲胺磷的标准溶液怎么存放?我们的是用甲醇稀释到50PPM在冰柜保存,6月配标,9月测试时响应挺高的,10月测试发现响应低了很多,仪器条件没变,这是为什么?
请问乙酰氯的GC分析方法极其标准?我们目前用GC-FID毛细管柱子,没有标准品,因为是化工企业,分析量大,请问这样分析的缺陷,用HPLC行不行?假如有其他微量杂质,是否还要用质谱仪?
最近做绿茶提取物甲胺磷,乙酰甲胺磷,对硫磷的检测,回收率做不出,甲胺磷乙酰甲胺磷才4-5%,对硫磷有70-80%。我使用的是安捷伦的6890N,检测器NPD,不分流进样,进标准品1.0ug/ml峰面积大概是300左右,但是不是很稳定。感觉问题出在前处理,分别用正己烷,乙酸乙酯,乙腈做过过加样回收,加2ml浓度1.0ug/ml。乙酸乙酯,乙腈提取浓缩后为粘稠红棕色液体,用无水硫酸镁和活性炭分散固相萃取后没有改善,正己烷提取后浓缩液无色。浓缩前进过仪器是可以测出来的,浓缩后没有了,回收液液没有,估计分解了。浓缩使用的是步其的平行定量浓缩仪,水浴45度,真空度250Mbar,冷凝水10-13度。我认为是浓缩步骤出的问题,大家帮分析一下,到底是哪里出的问题?有没有跟好的前处理分享一下,要方法简单的哦
急求标准对二氯苯、五氯化磷、乙酰氯、对甲苯磺酰胺、乙二醇甲醚、马来酸国标、化工标准、地方标准都行谢谢!![em0808]
4-氯乙酰乙酸乙酯的检测标准及检测方法N,N-二乙基乙二胺、N,N-二甲基乙二胺、N-乙基乙二胺系列产品,质量按Q/320801GHD001-2004标准执行、含量min.99.%
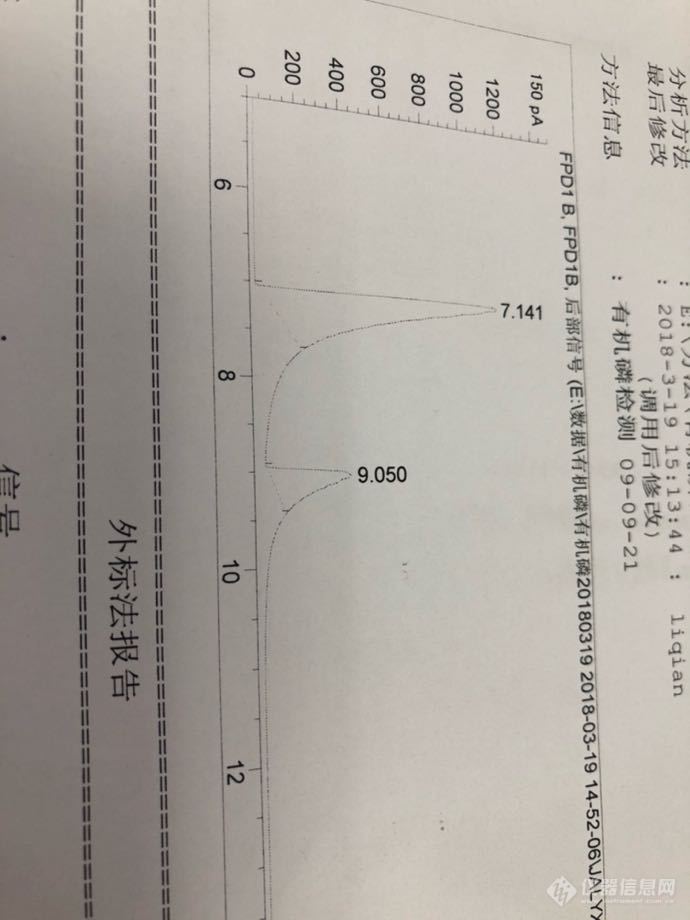
甲胺磷 乙酰甲胺磷拖尾严重…请问有啥解决办法吗?衬管也是新换的,柱头也割过一截了。方法也是之前用的。标准品也是新开的…请问有啥解决办法吗?[img=,690,920]http://ng1.17img.cn/bbsfiles/images/2018/03/201803191602167875_8255_3379628_3.png[/img]
本人现急需氯亚铂酸钾和二(乙酰丙酮)铂的分析方法或行业标准,那位大虾有相关资料,请上传或发到我的信箱[color=#DC143C]zhangfy03@126.com[/color],不胜感激!!!!
氯唑沙宗对照品和对乙酰氨基酚对照品的出峰时间分别是多久?
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
2008年9月22日,日本厚生劳动省发布食安输发第0922003号通知:近日,根据检疫所的监控检查结果,中国产新鲜胡萝卜中查出了乙酰甲胺磷。因此,日本厚生劳动省决定在对中国产胡萝卜及其加工品(限于简单加工)实行的命令检查的检查项目中追加乙酰甲胺磷。 违反事例: 1. 产品名称:新鲜胡萝卜 产 地:中国 申报数量及重量:2500箱,25000kg 检查结果:乙酰甲胺磷 0.02ppm(标准值:0.01ppm) 申报处:东京检疫所 违反确定日期:2008年5月28日 处理状况:全部保管中 2. 产品名称:新鲜胡萝卜 产 地:中国 申报数量及重量:2500箱,25000kg 检查结果:乙酰甲胺磷 0.03ppm(标准值:0.01ppm) 申报处:神户检疫所 违反确定日期:2008年9月19日 处理状况:1975箱已出库,525箱保管中国
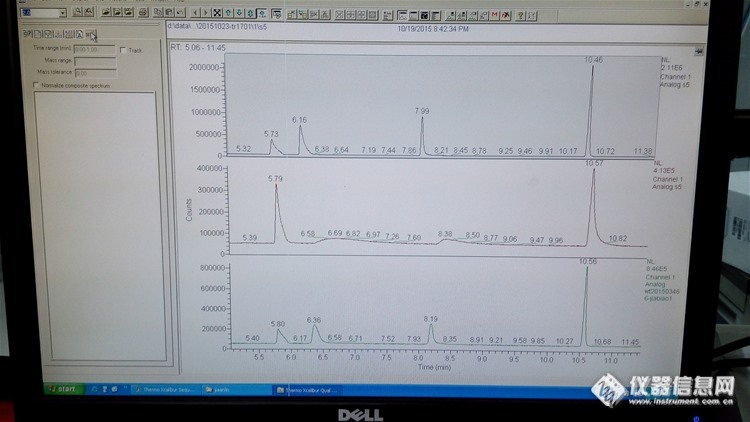
http://ng1.17img.cn/bbsfiles/images/2015/11/201511111107_573060_2547863_3.jpg如图,第一个谱图是1个月前做的标准曲线点,峰分别是敌敌畏,甲胺磷,乙酰甲胺磷,乐果,用的柱子是35柱第二个谱图是前天做的标准曲线点,其中甲胺磷,乙酰甲胺磷峰塌下去了第三个谱图是前天做的红枣加标样品,其中甲胺磷,乙酰甲胺磷峰很正常。如今往红枣样品上机液(四个峰均不出峰)添加混标液,得出的谱图和第三个一样;重新配了混标,甲胺磷和乙酰甲胺磷也是超级拖尾,但是一旦把混标加入样品液里面,两个出峰都很正常求解决方法(已经切割过柱子前端,进样瓶都是新的,除了溶液一个是丙酮定容的样品上机液,一个是色谱纯丙酮不一样外,其他条件都一样)
用串接质谱(包括单极)检测乙酰甲胺磷时离子碎片比不稳定,也就是说走标准,离子碎片比例都不一致。为什么?大家有没有发现过。希望专家给予答复。当然19648中也没有乙酰甲胺磷这个目标物。
请教:复方乙酰水杨酸钠含量用什么方法测定?
请教各位:元素分析用的标准物质乙酰苯胺的定值不确定度是从哪里得知的?我们实验室里的乙酰苯胺标准物质是从上海市计量测试技术研究院买的,在标准物质证书上只有百分理论值,并没有列出不确定度。
测点药片中的乙酰水杨酸时,为什么1mol乙酰水杨酸只消耗2molNaOH?酚羟基不与NaOH反应吗?

最近在做对乙酰氨基酚片溶出度实验,首先测定对乙酰氨基酚的标准曲线,给出的方法如下,仪器为TU1901,对乙酰氨基酚为对照品。[img]http://ng1.17img.cn/bbsfiles/images/2008/05/200805302132_91355_1630080_3.jpg[/img]以上方法是一本教材上的方法,我们严格按照上述方法,溶剂也采用0.04%NaOH,定量测定结果如下,结果很不理想,重配标液后再测,结果同样不理想,后对每一标液做光谱扫描,标1至标4最大吸收波长均在257nm左右,标5和标6最大吸收波长则移动到243nm。 [img]http://ng1.17img.cn/bbsfiles/images/2008/05/200805302137_91356_1630080_3.jpg[/img]经过反复查找资料和分析,推断标5和标6母液取样量大,破坏了溶剂的PH值,致使最大吸收波长发生迁移,遂将溶剂改为0.4%NaOH,重新配制标准溶液进行测定,如果如下图,相关系数很好。 [img]http://ng1.17img.cn/bbsfiles/images/2008/05/200805302150_91359_1630080_3.jpg[/img][color=#DC143C][size=4]总结两点:一、溶液酸碱度对对乙酰氨基酚定量测定有较大影响,实验时应消除PH影响。二、教材及文献的实验方法应动手验证,否则也会有差错。[/size][/color]
问题:.请问甲维盐或 乙酰甲胺磷的制剂用什么方法来做?有标准号吗?
如题,求助两个标准。乙酰乙酸甲脂、对甲苯磺酸相关标准。Email: paigu064@163.com
[size=3][size=4]我需要做乙酰丙酸的标准曲线,看别的文章后,按他写的方法试过,完全不是他说的结果,请问哪位做过乙酰丙酸的标准曲线,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],可否将方法相告!不胜感激![/size][/size]







 我要推广仪器
我要推广仪器
 下载APP
下载APP