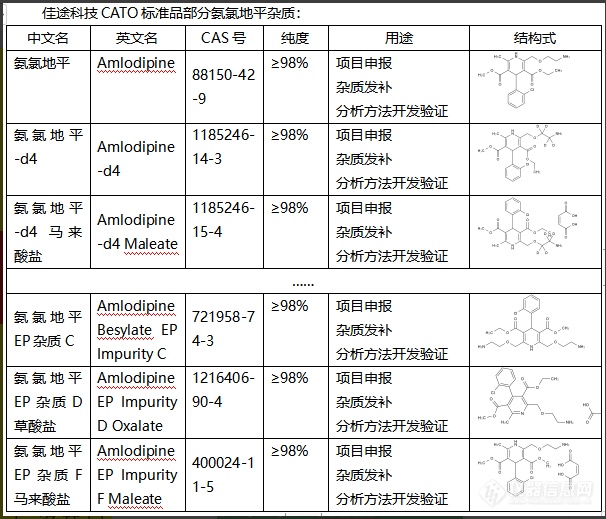
氨氯地平是一种常用的抗高血压药物,对于其中的杂质,氨氯地平杂质其作用主要体现在以下几个方面:1. 质量影响:杂质会影响氨氯地平的纯度和稳定性,可能会导致药品质量下降。2. 安全性影响:杂质可能会产生一些未知的副作用和毒性反应,影响药品的安全性。3. 药效影响:杂质可能会干扰氨氯地平的药效,使得药物的治疗效果降低。4. 法规因素:食品药品监管部门对药品中的杂质有严格的限制标准,过多的杂质可能会导致药品不能上市。CATO标准品对于氨氯地平这类药物的生产,控制和降低杂质的含量是非常注重的。[img=,606,519]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041354066781_2922_6381668_3.png!w606x519.jpg[/img]
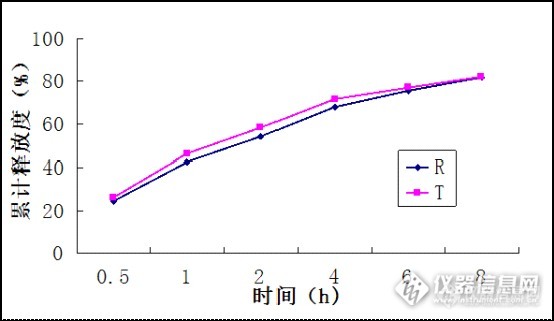
各位大神想请教一个问题,本人做苯磺酸氨氯地平的方法学试验,样品是儿童铁锌钙口服液(保健品),标准是国家食药监总局药品补充检验方法和检验项目批准件(降压类中成药和辅助降血压类保健食品)第2011008号。一开始是几个地平一起做的(氨氯、尼群、非洛、硝苯),做了三个加标水平(10PPb、40ppb、80ppb)其他三个都没有问题,但是氨氯地平的三水平的回收率都非常高,超200%了,做了样品中是没有的,然后又单独配了氨氯地平的标曲和重做了回收率,这次比混标稍微好一点,但是三水平的回收率还是偏高,分别是160%、160%、130%,请问有遇到过类似情况的吗?是因为样品的问题吗?(就是说氨氯地平跟样品中的某些物质发生反应?我查看了图谱,标准的峰比较正常,但是样品加标的峰会比较胖一些),请各位大神指点!谢谢
各位前辈,今日做苯磺酸氨氯地平片,看到新药转正标准上鉴别有红外鉴别一项,分别用氯仿、甲醇、无水乙醇提取后做红外图谱,但是和对照图谱集对不上哦,这是什么情况,有作过的朋友指点一下,到底有什么提取,怎样提取出来,感谢!
各位前辈,今日做苯磺酸氨氯地平片,看到新药转正标准上鉴别有红外鉴别一项,分别用氯仿、甲醇、无水乙醇提取后做红外图谱,但是和对照图谱集对不上哦,这是什么情况,有作过的朋友指点一下,到底有什么提取,怎样提取出来,感谢![color=#f10b00]见此贴:[/color][url]http://bbs.instrument.com.cn/shtml/20091027/2177496/[/url]
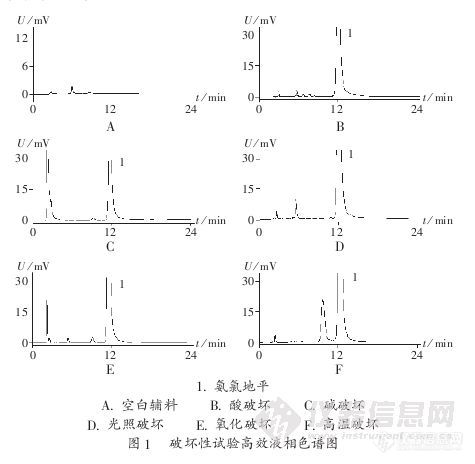
作者:秦书德 马晓伟 陈静 郭兴杰作者单位沈阳药科大学,摘 要:目的建立检查苯磺酸氨氯地平有关物质的反相高效液相色谱法。方法采用Diamonsil C18色谱柱(200 mm×4.6 mm,5μm)进行分离,流动相为甲醇-30 mmol/L磷酸二氢钾溶液(60∶40),流速1.0 mL/min,检测波长238 nm。结果所用色谱条件能很好地分离氨氯地平及其降解产物,氨氯地平质量浓度在0.71~3.57μg/mL范围内与峰面积线性关系良好。结论反相高效液相色谱法专属性强、准确、灵敏,可用于苯磺酸氨氯地平片中有关物质的检查:http://ng1.17img.cn/bbsfiles/images/2012/08/201208201631_384755_2379123_3.jpg
苯磺酸氨氯地平 2015年版二部药典上添加了有关物质I 是薄层色谱法 在紫外灯254nm和365nm下检视 大家觉着是分别开这两个波长的灯还是一起开?
药品质量标准中鉴别项目设置的几点考虑 审评三部 张哲峰 摘要:本文简要介绍了药品质量标准中常用的几种鉴别方法,并对常用鉴别方法的优势和局限进行了分析,针对鉴别项目设置中需注意之处提出了一些看法。 关键词:质量标准 鉴别项目 药品质量标准中鉴别是用以判定某已知药品的真伪而不是对未知药物进行结构确证,所以鉴别方法应以专属性好、简便易行为宜,尤其能将结构相似的同类药品加以区别为主要考虑因素。如新鱼腥草素钠及制剂标准中仅用化学法和UV法作鉴别,难以与结构类似物鱼腥草素钠及制剂相区分,质量标准不具备应有的专属性,可能给此后的市场监督造成混乱。 常用的鉴别方法包括色谱法、光谱法、化学法和生物学方法等,可根据药品具体特点加以选用。 色谱法(TLC法或HPLC法)利用不同物质在不同色谱条件下,各自色谱行为(比移值或保留时间)的不同,与对照品在相同色谱条件下进行色谱分离,比较其色谱行为的一致性,来鉴别药品的真伪。这类方法的运用使得结构相似化合物、同系物等的区分变得简单易行。HPLC法虽然主要用于定量,但如果运用得当,尤其在含量测定或有关物质项下已采用本法的情况下,利用对照品与供试品保留时间相同的特性作为鉴别依据,不必专门增加实验以提高鉴别的专属性,是非常可取的。值得注意的是色谱系统的稳定性要好,同一物质不同进样时保留时间的重现性必须有保证。这就要求流动相与固定相相匹配,C18链在水相环境中不易保持伸展状态,故在C18柱的反相色谱系统中,流动相有机溶剂比例通常不应低于5%,否则C18链的随机卷曲将造成色谱系统不稳定导致组份保留值波动,不利于此种鉴别。即便如此,在实际操作中有时依然能遇到同一物质在完全相同的色谱系统中保留时间不一致的情况,尤其梯度洗脱时此种现象更为常见。药典中对保留时间的一致性未予具体规定,此时,操作中可增加供试品溶液与对照品溶液等量混合,进样后出现单一色谱峰作为鉴别依据,可以弥补该法之不足,此操作可列入质量标准。在含量和有关物质未采用HPLC法的情况下,一般不单独采用本法作鉴别。 TLC法除色谱行为外,还可将斑点颜色作为鉴别依据,可由两个因素把握供试品与对照品的同一性,而且简便易行,堪称一个很好的鉴别方法。但由于薄层板质量、边缘效应等因素的影响,实际操作中有时也会遇到同一物质在同一块薄层板上的Rf值不一的情况,可比照HPLC的情况,操作中增加供试品溶液与对照品溶液等量混合,点样后出现单一斑点作为鉴别依据,此点在2005年版药典中已有体现。也有人提议明确Rf值偏差不超过5%,作为鉴别要求,但其可行性有待考察。单独使用TLC鉴别时,要有色谱系统适应性试验内容,如要求几种结构相似化合物的混合溶液色谱展开后应显示相应的几个斑点或最难分离物质对能够分开的情况下,供试品溶液与对照品溶液主斑点的颜色与位置应一致。 在中国药典2005年版中,对TLC鉴别法在斑点的颜色与位置明确规定的基础上对斑点大小也做出明确要求:供试品与同浓度对照品溶液颜色与位置应一致,斑点大小应大致相同;或供试品与对照品等体积混合,应显示单一,斑点紧密;或供试品溶液的主斑点与上述混合溶液的主斑点的颜色与位置一致,大小相似;或选用与供试品化学结构相似药物对照品,两者的比移植应不同(例如芬布芬与酮洛芬,地塞米松磷酸钠与泼尼松龙磷酸钠,醋酸氢化可的松与醋酸可的松,泼尼松龙与氢化可的松,甲睾酮与睾酮,左旋多巴与酪氨酸);或上述两种溶液等体积混合,应显示两个清晰分离的斑点。 光谱法中IR法因可反映较多的结构信息,在组份单一、结构明确的原料药鉴别中作为首选, 药物存在多晶型现象又无可重复转晶方法时一般不采用此法,但如果药物存在多晶型现象,且需鉴别其有效晶型,IR图谱可以反映其有效晶型特点时,本法又是一种有效而简便易行的鉴别方法。制剂中则因辅料影响、提取过程可能导致晶型变化而一般不采用IR法,而采用所受影响因素较少的UV法。 常用的UV鉴别方法有:测定最大吸收波长,或同时测定最小吸收波长;规定一定浓度的供试液在特定吸收波长(最大吸收或最小吸收)处的吸收度;经化学处理后,测定其反应产物的吸收光谱特征;规定几个特定吸收波长及其吸收度比值(峰-峰、峰-谷、谷-谷);规定几个特定吸收波长及其吸收系数。因末端吸收所受影响因素较多,UV法鉴别时,一般不宜用220nm以下波长的吸收特性作鉴别;因反映的结构信息少,一般也尽量不用单一吸收峰作鉴别依据;为提高专属性,可将上述几个方法结合起来使用。 化学鉴别法一般是特定官能团或特定结构化合物的特性反应,与其它鉴别方法结合使用,可以使得鉴别的专属性更加突出。化学鉴别法具有专属性较强、反应迅速、现象明显的特点才有使用价值。包括在适当条件下产生颜色、荧光,发生沉淀反应或产生气体等现象。 1.呈色反应:即向供试品溶液中加入适当试剂,在一定条件下发生化学反应,生成易于观测的有色产物。常见的反应类型有:[/c
本人正在做中药的生物碱提取,我之前看了资料,对样品进行了初步提取(最后一步是氯仿萃取),然后选择改良碘化铋钾、碘-碘化钾和磷钼酸进行定性,但是当我把这三种试剂分别滴到样品之后,问题来了:1、 理论说这三种试剂如果有阳性反应的话,是呈橘红色、棕黄色的,但我滴下去后,出现的是分层了,而没有沉淀出现。为什么大家都说加进去后会有沉淀呢?样品的溶剂是氯仿,而沉淀试剂的溶剂是稀冰醋酸,这两者是不相溶的,那何来的反应出现沉淀呢?2、我要提取的生物碱,目前还没有标准品可以购买,对于这种没有标准品的生物碱,要拿什么做对照品呢?请大家多多指教。我查了文献,也有其他人在做同样这种情况的实验(没有标准品的生物碱提取),但他们是用其他的生物碱作为对照品来鉴别的,可以这样的吗?这样做的依据是什么呢?好迷茫啊,希望大家多多回复啊!!!!谢谢了!
本人正在做中药的生物碱提取,我之前看了资料,对样品进行了初步提取(最后一步是氯仿萃取),然后选择改良碘化铋钾、碘-碘化钾和磷钼酸进行定性,但是当我把这三种试剂分别滴到样品之后,问题来了:1、 理论说这三种试剂如果有阳性反应的话,是呈橘红色、棕黄色的,但我滴下去后,出现的是分层了,而没有沉淀出现。为什么大家都说加进去后会有沉淀呢?样品的溶剂是氯仿,而沉淀试剂的溶剂是稀冰醋酸,这两者是不相溶的,那何来的反应出现沉淀呢?2、我要提取的生物碱,目前还没有标准品可以购买,对于这种没有标准品的生物碱,要拿什么做对照品呢?请大家多多指教。我查了文献,也有其他人在做同样这种情况的实验(没有标准品的生物碱提取),但他们是用其他的生物碱作为对照品来鉴别的,可以这样的吗?这样做的依据是什么呢?好迷茫啊,希望大家多多回复啊!!!!谢谢了!
有药品/食品用复合塑料袋的红外鉴别标准图谱吗?
食品安全质量鉴别与国家检验标准全书[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=114963]食品安全质量鉴别与国家检验标准全书[/url][img]http://ng1.17img.cn/bbsfiles/images/2008/10/200810271936_114964_1634467_3.jpg[/img]
增效联磺片为磺胺类抗菌消炎药的新型复方制剂,每片含磺胺甲基异(口恶)唑200mg、磺胺嘧啶200 mg、甲氧苄氨嘧啶80 mg,各地方标准均有收载,对前两种成分以纸色谱法鉴别,而对甲氧苄氨嘧啶则另行鉴别。本文以薄层色谱法同时鉴别磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶3种成分,专一性强,斑点明显,操作简便,结果较为满意。1 仪器与试药 三用紫外线分析仪(上海顾村电光仪器厂),硅胶GF254薄层板(10cm×20 cm,自制);磺胺甲基异(口恶)唑、磺胺嘧啶和甲氧苄氨嘧啶对照品(中国药品生物制品检定所);增效联磺片(市售品);硅胶GF254(青岛海洋化工厂生产,化学纯);其它试剂均为分析纯。2 溶液的配制2.1 单一对照品溶液 分别精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液分别制成0.5 mg/mL磺胺甲基异(口恶)唑、0.5 mg/mL磺胺嘧啶、0.2 mg/mL甲氧苄氨嘧啶的单一对照品溶液。2.2 混合对照品溶液 精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液制成? mL含磺胺甲基异(口恶)唑0.5 mg、磺胺嘧啶0.5 mg和甲氧苄氨嘧啶0.2mg的混合对照品溶液。2.3 样品溶液的配制 取供试品细粉适量(约相当于磺胺甲基异(口恶)唑50mg),加50 %丙酮溶液100 mL,振摇使溶解,过滤,滤液作为供试品溶液。
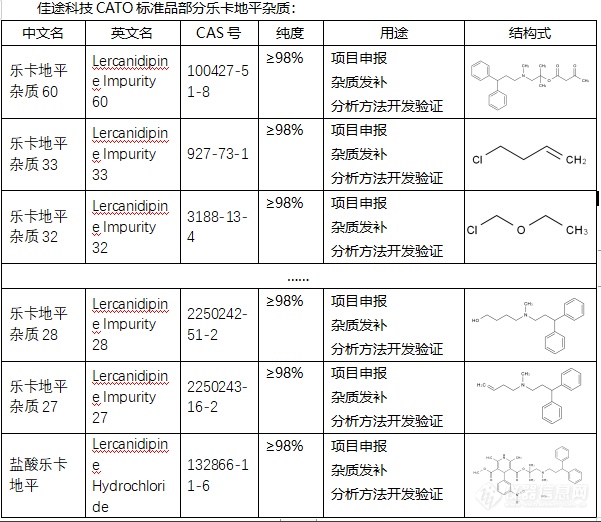
乐卡地平杂质的作用是多方面的,并且主要取决于它们的类型和含量。1. 降低药效:如果杂质的含量过高,可能会降低乐卡地平的药效。例如,杂质可能会与乐卡地平的活性成分竞争,降低其与体内靶点的结合能力,从而降低其药效。2. 引发副作用:某些类型的杂质可能会引发副作用,例如过敏反应、肝脏损伤等。这通常取决于杂质的类型和含量,以及患者的个体差异。3. 质量控制:在药品制造过程中,杂质也被用作一种质量控制的手段。通过测量杂质的含量,可以评估药品的纯度和质量,以确保药品的安全性和有效性。CATO标准品药品中杂质的含量都会严格控制在一定范围内,以确保药品的安全性和有效性。药品在上市之前,必须经过严格的质量检测,以确保其杂质含量符合法定标准。[img=,601,522]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041448095358_8809_6381668_3.png!w601x522.jpg[/img]

HPLC法测定西尼地平的有关物质西尼地平(Cilnidipine)为一新型抗高血压药,其通过L-型钙通道长时间地抑制钙内流,具有长时间持续扩张血管作用,还可预防卒中和降低死亡率并减少肾、心、血管并发症,具有良好的发展前景。有关西尼地平药理作用的文献较多,但其质量分析方法的研究报道较少,本文采用反相高效液相色谱面积归一化法,对西尼地平合成工艺中各步中间体及可能产生的有关物质予以分离检测。 具体方法如下:(一)色谱条件:色谱柱:十八烷基硅烷键合硅胶柱(Xtimate C18),250×4.6mm,5um流动相:0.03mol/L磷酸氢二钠溶液(用磷酸调节pH为4.0)-甲醇-乙腈(40:35:25)检测波长:240nm流速:1.0ml/min进样量:20ul系统适用性试验要求:理论塔板数按西尼地平峰计算应不低于3000;在供试品图谱中西尼地平与相邻杂质峰的分离度应符合规定。(二)供试品溶液:称取本品约10mg,加甲醇溶解并稀释至50.0ml,制成每1ml含0.2mg的溶液,作为供试品溶液。(临用新制) (三)测定:按照高效液相色谱仪操作规程准备仪器,平衡后取空白溶液20ul注入液相色谱仪,再取供试品溶液20ul注入液相色谱仪,记录色谱图至主成分峰保留时间的4倍。色谱图如下:http://ng1.17img.cn/bbsfiles/images/2012/12/201212231401_414537_2583865_3.jpg结果计算:供试品溶液的色谱图如有杂质峰,按面积归一化法计算:单个杂质峰面积不得大于所有峰面积总和的0.2%;各杂质峰面积的和不得大于所有峰面积总和的0.5%。注意:供试品溶液配制应避光操作。
中间品的性状,鉴别在企业内控质量标准,操作规程,记录,报告中还用编写吗?有这方面的指导规范吗?
请问大家用GG做辅料鉴别的吗?所用的标准试剂都是买哪里的啊,像甲醇、乙醇、二氯甲烷、乙酸乙酯这类辅料,我们买的是不同厂家的来相互鉴别,总觉得觉得这种做法有问题,请问有专门卖这种标准试剂的公司吗?
各位老师都是做食品安全的高手,想请教一下,除了眼观手摸意外,有没有知道检测水果打蜡的方法?或者如何鉴别用的是食用石蜡还是工业石蜡??国家目前有没有出具相应的标准?
最新食品安全质量鉴别与国家检验标准全书 http://www.instrument.com.cn/download/search.asp?keywords=%D7%EE%D0%C2%CA%B3%C6%B7%B0%B2%C8%AB%D6%CA%C1%BF%BC%F8%B1%F0%D3%EB%B9%FA%BC%D2%BC%EC%D1%E9%B1%EA%D7%BC%C8%AB%CA%E9&sel=title&SN=&Submit=%C1%A2%BC%B4%B2%E9%D1%AF
紫外分光光度法测定硝苯地平缓释片释放度高血压是当今世界最常见的心血管疾病之一,也是心脑血管疾病的主要危险因素。硝苯地平是二氢吡啶类的钙通道阻滞剂,临床广泛用于冠心病、心绞痛及原发性高血压等疾病的治疗。目前,其普通制剂在国际上已被逐步淘汰,取而代之的是硝苯地平长效缓、控释制剂。本文开发快速测定硝苯地平缓释片体外释放度的方法,并测定其释放曲线,为其质量标准的制定提供依据,并为其体内评价提供数据支持。1仪器与材料仪器:UV-2450紫外分光光度计(日本岛津);BS 1101电子天平( 北京赛多利斯天平有限公司生产);RCZ-8M型智能溶出仪(天津天大天发科技有限公司)。材料:硝苯地平对照品(中国药品生物制品检定所);十二烷基硫酸钠(SDS,汕头市西陇化工有限公司) ,其他试剂均为分析纯。2方法和结果2.1标准储备液精密称定硝苯地平对照品50 mg于100 mL容量瓶中,先加入少量甲醇溶解,以无水乙醇稀释至刻度,作为储备液。2.2最大检测波长精密移取2mL储备液于50mL容量瓶中,用0.5%SDS水溶液配置成一定浓度溶液,以相应的溶剂作空白,按紫外分光光度法在200~400 nm的波长范围内进行紫外扫描,确定紫外最大吸收波长。扫描结果如图1所示。http://ng1.17img.cn/bbsfiles/images/2013/11/201311300747_480065_1903863_3.png图1 硝苯地平在0.5%SDS中的紫外扫描图由图1可知,硝苯地平在236 nm和333 nm有最大吸收,根据峰形选择333nm。2.3空白辅料干扰实验精密称取处方量的辅料18.50mg,置于100 mL容量瓶中,用0.5%SDS水溶液稀释定容,按紫外分光光度法在200~400 nm的波长范围内进行紫外扫描。扫描结果见图2。http://ng1
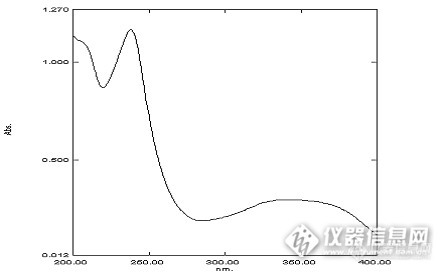
紫外分光光度法测定硝苯地平在不同介质中的饱和溶解度高血压是当今世界最常见的心血管疾病之一,也是心脑血管疾病的主要危险因素。目前全球有高血压患者6亿人。硝苯地平是目前临床上治疗高血压和心绞痛首选药物之一,其普通制剂在国际上已被逐步淘汰,取而代之的是硝苯地平长效缓、控释制剂。本文开发快速测定硝苯地平饱和溶解度的紫外分光光度法,并检测硝苯地平在不同介质中的饱和溶解度,为其缓控释制剂的开发提供数据支持。1仪器与材料仪器:UV-2450紫外分光光度计(日本岛津);SHA-C水浴恒温振荡器(金坛市白塔金昌实验仪器厂)。材料:硝苯地平(山西临汾宝珠制药有限公司);十二烷基硫酸钠(SDS,汕头市西陇化工有限公司) ,其他试剂均为分析纯。2方法和结果2.1.标准储备液的配备精密称定硝苯地平对照品50 mg于100 mL容量瓶中,先加入少量甲醇溶解,以无水乙醇稀释至刻度,作为储备液。2.2最大检测波长的确定精密移取2mL储备液于50mL容量瓶中,用0.5%SDS水溶液配置成一定浓度溶液,以相应的溶剂作空白,按紫外分光光度法在200~400 nm的波长范围内进行紫外扫描,确定紫外最大吸收波长。扫描结果如图1所示。http://ng1.17img.cn/bbsfiles/images/2013/11/201311300745_480064_1903863_3.png图1 硝苯地平在0.5%SDS中的紫外扫描图由图1可知,硝苯地平在236 nm和333 nm有最大吸收,根据峰形选择333nm。2.3 标准曲线的建立分别精密移取标准储备液于适宜大小的容量瓶中,用0.5%SDS水溶液稀释定容,配制成45μg/mL[fo
标准品:水质氨氮浓度:0.778mg/L安瓿瓶包装20mL问:在证书上面其中有一段为 - 本环境标准品应按以下程序稀释后方可使用:临用前小心打开安瓿瓶,用10mL干燥洁净移液管从安瓿瓶中准确量取10mL浓样至250mL容量瓶中,用纯水稀释定容至刻度,混匀后立即使用。请问其意思是稀释定容250mL才是证书上写的0.778mg/L浓度吗?
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做农药标准品时没有出峰,而且杂质峰较多,更换条件后8个物质的混标只出了6个峰,而且很小,并有数个有氯碎片。是不是标准品失效?
蜂蜜检测仪是一种专门用于鉴别蜂蜜真假的设备,它基于先进的技术手段,能够准确、快速地分析蜂蜜中的成分,从而判断其真伪。 蜂蜜检测仪的操作步骤 准备样品:从待检测的蜂蜜中取出适量样品,确保样品具有代表性。同时,准备好标准蜂蜜样品作为对照。 设置检测参数:根据蜂蜜检测仪的说明书,设置合适的检测参数,包括检测波长、测量时间等。这些参数的设置对于测量结果的准确性至关重要。 进行测量:将蜂蜜样品和标准样品分别放入蜂蜜检测仪的检测通道中,启动仪器进行测量。仪器会自动对样品中的蔗糖、果糖、葡萄糖以及甲基糠醛等成分进行分析,并给出相应的数据。 分析测量结果:测量完成后,可以通过仪器的显示屏查看每个样品的测量数据。将待检测蜂蜜的数据与标准蜂蜜的数据进行对比,如果两者差异较大,则很可能是假蜂蜜或者掺假蜂蜜。 蜂蜜检测仪能够准确鉴别蜂蜜的真假,主要体现在以下几个方面: 成分分析:蜂蜜检测仪能够精确测量蜂蜜中蔗糖、果糖、葡萄糖等关键成分的含量,从而判断蜂蜜是否纯正。 掺假检测:通过对比待检测蜂蜜与标准蜂蜜的数据,可以判断蜂蜜是否存在掺假行为,如添加糖精、水等。 快速高效:蜂蜜检测仪能够在短时间内完成大量样品的检测,提高了检测效率。 总之,蜂蜜检测仪是一种有效的工具,能够帮助我们准确鉴别蜂蜜的真假。通过科学、规范的操作和维护,可以充分发挥其鉴别能力,保障消费者的权益。 在农产品收购环节,可以使用这些设备进行快速筛查,避免收购到农药残留超标的农产品。在食品加工前,对原料进行农药残留检测,确保原料的安全性,防止因原料问题导致的食品安全事故。 在食品加工前,对原料进行农药残留检测,确保原料的安全性,防止因原料问题导致的食品安全事故。 在销售前对农产品进行快速检测,确保销售的农产品符合食品安全标准,保障消费者的健康。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/07/202407081453095005_4648_6238082_3.jpg!w690x690.jpg[/img]
用气相-ECD进有机氯标准品混标,原来各种标准品都很好,但最近里面唯独百菌清没有出峰了,其他都很好,不知道怎么回事?
[em09506]请问谁可以提供非洛地平氧化产物对照品?谢谢!
以V氯仿∶V甲醇∶V二氯甲烷∶V正己烷=2.6∶1.2∶1∶5为展开剂, 建立了薄层色谱扫描法测定痕量药物尼莫地平的新方法。 其Rf值为0.48。 扫描波长为365 nm, 该法的最低检出限为0.005 μg,相对标准偏差为2.94%, 工作曲线的线性范围为0.005~1 μg。 用该法测定了加有尼莫地平的血清和尿样, 尼莫地平的平均回收率为96.7%~103%。关键词 尼莫地平, 紫外薄层色谱法 尼莫地平(nimodipine)为一钙离子拮抗剂, 能有效地调节细胞内钙的水平, 具有抗缺血和抗血管收缩作用[1], 是近年来治疗高血压和脑血管疾病的一种新药, 其结构式如右所示。 有关该药的测定方法, 曾报道过的有高效液相色谱法[2]、 分光光度法[3], 但用紫外薄层色谱扫描法测定尼莫地平至今未见文献报道。 本文首次采用紫外薄层色谱法, 以氯仿-甲醇-二氯甲烷-正己烷为展开剂, 对尼莫地平的测定进行了研究。 该法具有简单、 快速、 灵敏、 准确的特点, 我们成功地做了血清和尿样中不同浓度的加标回收实验, 结果令人满意。 该法可用于临床作为测定尼莫地平血药及尿药浓度的一种简单、 有效的新方法。1 实验部分1.1 仪器与材料 CS-9000双波长薄层色谱扫描仪(日本岛津); 毛细管定量点样器(美国Drummond); UV-1型紫外分析仪(上海顾村电光分析仪器厂); 硅胶GF254 板(青岛海洋化工厂); 尼莫地平(山东新华制药厂提供)。 其它试剂均为分析纯以上规格。1.2 实验方法 用1 μL定量毛细管点样, 标样与试样点于同一板上 待溶剂挥发后, 放入盛有展开剂的层析缸中, 用蒸汽预吸附3~5 min, 然后用展开剂展开, 展开剂为V氯仿∶V甲醇∶V二氯甲烷∶V正己烷=2.6∶1.2∶1∶5, 展开到距板上端1 cm处, 展距9 cm 取出板, 待溶剂挥发干净, 置于紫外分析仪, 在254 nm波长下可观察到样品暗红色斑点, 尼莫地平的Rf值为0.48 然后用薄层色谱扫描仪扫描, 以外标两点法定量。 紫外薄层扫描条件: 以尼莫地平光谱λmax=365 nm为测定波长,锯齿扫描, 数据累加4, 数据平滑11,高灵敏度。2 结果与讨论2.1 展开剂的选择 我们根据Glajch三角形最优化法[4]及参照Snyder溶剂参数法[4]对展开剂的组成及配比进行了选择,确定了以氯仿-甲醇-二氯甲烷-正己烷作为四元混和展开剂,其配比为V氯仿∶V甲醇∶V二氯甲烷∶V正己烷=2.6∶1.2∶1∶5 。
[table][tr][td][size=3]中药化学对照品研究是"中药现代化研究与产业化"项目中 "建立中药系列标准规范"课题的重要内容,也是中药实现国际化、现代化的关键。进行常用中药材质量标准及化学对照品研究,应用现代分离、分析手段制备化学对照品,并建立符合中医药特点且达到国际通用标准的中药材质量控制及评价方法,为制定中药材及中成药质量标准体系提供保证。为规范中药化学对照品研究专题的研究内容及验收指标,特制定中药化学对照品研究指导原则及验收标准,供专题承担单位和研究人员参照执行。 一、目的及意义 中药材是中药研究与开发的基础,历来倍受重视。目前,中药材的生产、采收、饮片加工及质量评价有待进一步完善与规范,在中药现代化、国际化进程中,首先必须从中药材的质量抓起。中药化学对照品的研究对中药材及中成药质量标准建立至关重要。中药化学对照品研究专题的设置目的是通过现代科学方法,制备并提供能够满足科研和生产中所需的中药化学对照品,进而为建立具有中医药特点的中药材质量标准规范体系及开展中药现代化研究提供技术保障。制定科学规范的中药材质量标准对于确立我国传统医药大国的主导地位、促进中药进入国际市场并争取更多的市场份额,具有十分重要的战略意义。 二、指导原则 随着科学技术的发展,在现阶段中药研究水平基础上,对中药材的有效成分或指标性成分进行研究,参考现代医药研究的国际规范,吸收各国传统医药应用和管理经验,完善和建立符合中医药特点的中药材质量标准体系。 1. 制定中药材质量标准,首先必须以中医药理论为基础,充分考虑中医药的特点。中医临床用药注重整体观点,这不仅是中医药几千年来的用药习惯,也是中医药区别与西医药的重要方面。大量临床验证及药理实验均证明,单一成分或组分与单味药的作用往往是不能等同的,这是因为中药材所含成分复杂,其药效可能是各成分的综合作用。因此,仅用某一成分作为指标,来衡量该药材的质量,不能体现其内在品质。因此,建立主产区药材的指纹图谱,并在建立单指标成分含量测定基础上,探讨多指标成分综合评价体系是非常必要的。 2.固定药材的植物来源,多植物来源的药材分别进行研究,制定能反映其内在质量的规范标准。 3.应在国家法定标准的基础上参考国际上有关草药质量控制方法,建立科学、可控的中药材质量标准。 三、研究内容 应用现代色谱及分析手段,对中药材的化学成分进行提取、分离、结构测定,确定其有效成分或指标性成分,制备中药化学对照品。在来源、形状、鉴别、检查、浸出物、有效成分或指标性成分及定性定量分析、主产地药材指纹图谱、重金属含量及农药残留量检测等方面进行规范化研究,参照现行中国药典制定其质量标准。 ㈠、中药化学对照品研究内容 1.对中药材的化学成分及药理作用进行文献调研。 2.通过对所选品种的化学成分及药理研究,确定该药材的有效成分,无法确定有效成分的,可选用指标性成分。 3.对照品的制备工艺研究,包括可以较大量提取、分离该对照品的方法及相应条件和详细的工艺流程研究。 4.对照品结构、性质研究: (1) 中英文名称,分子式,结构式 (2) 性状及理化常数:提供结晶溶剂、晶形、溶解性能、熔点或沸点、旋光度等数据。 (3) 薄层色谱鉴别:包括所用色谱条件、显色条件、Rf值及彩色照片。 (4) 结构测定:对已知化合物,其数据及图谱与文献值或图谱一致;如不一致,按未知物标准处理。未知物要求提供足以确证其结构的化学及物理学数据(IR、UV、 NMR、MS等)。 (5) 纯度及其检查方法。 (6) 含量测定:提供含量测定的方法、数据及有关图谱。 (7) 初步稳定性:根据化合物的理化性质,确定储藏条件。 ㈡、中药材质量标准的研究内容 1.对药材的资源、生产及市场情况进行考察,尽量提供植物来源、资源分布、市场品种及其主产地情况,尽量选择道地药材品种进行研究。 2.对药材样品进行基源鉴定,同时注明产地、采收时间及加工方法。 4.主产地药材指纹图谱研究:研究主产地药材指纹图谱,优先选择色谱法,对不同产地商品进行分析,确定共有峰,尽可能获得较多的色谱峰作为该药材的特征控制其质量。 5.中药材鉴别方法研究:鉴别方法的研究包括经验、显微、理化及色谱鉴别。以有效成分或指标成分/对照药材为参照,提供鉴别特征。鉴别方法必须具有专属性和重现性。 6.重金属和农药残留物的监测方法及分析:用现代化分析技术进行重金属和农药残留物的检测分析。并参照如下标准执行:重金属总量≤200mg/kg;铅(Pb)≤5.0g/kg;镉(Cd)≤0.3mg/kg; 汞(Hg)≤0.2 mg/kg; 铜(Cu)≤20.0mg/kg ;砷(As)≤2.0 mg/kg。农药残留量 六六六(BHC)≤0.1mg/kg ;DDT≤0.1mg/kg;五氯硝基苯(PCNB)≤0.1mg/kg;艾氏剂(Aldrin)≤0.02mg/kg。 7.毒剧成分应制定限量范围。 8.制定中药材质量标准草案及起草说明。 四、中药化学对照品研究技术要求及验收指标 1.提供国内外对该药材的有效成分或化学成分的详细研究情况报告。 2.提供该药材的植物来源、资源分布、市场品种及其主产地情况。 3.药材样品的基源鉴定,同时注明产地、采收时间及加工方法。 4.选择、确定对照品的实验依据。 5.要求提供有效成分或指标性成分下列数据: ⑴ 中英文名称,分子式,结构式; ⑵ 性状及理化常数:提供结晶溶剂、晶形、溶解性能、熔点或沸点、旋光度等数据; ⑶ 中药化学对照品的提取制备工艺流程:提供标准品的详细提取、分离及纯化方法及收率; ⑷ 薄层色谱检查,包括所用吸附剂、溶剂系统、显色剂及Rf值,提供彩色照片; ⑸ 纯度及其检查方法:纯度检查可依所用的色谱类型,如为薄层色谱法、点样量应为所适用检验方法点样量的10倍量,选择三个以上溶剂系统展开,并提供彩色照片; ⑹ 含量测定:提供含量测定的方法、数据及有关图谱。提供含量测定用的对照品纯度应在98%以上,供鉴别用的对照品纯度应在95%以上; ⑺ 初步稳定性:根据其稳定性确定储藏条件; 6.鉴别方法:以有效成分或指标成分或对照药材为参照,提供鉴别特征。鉴别方法必须具有专属性和重现性。 7.药材中有效成分或指标性成分的含量测定方法:要求优先考虑高效液相色谱法等目前较常用方法。可按新药研究中有关含量测定要求进行。 8.药材中毒剧成分的限量范围及依据。 9.主产区药材指纹图谱:优先选择色谱法。对不同产地至少10批样品进行分析,确定共有峰,尽可能获得较多的色谱峰作为该药材的特征控制其质量。 10. 重金属和农药残留物的检测分析及结果。 11.参照现行药典制定的中药材质量标准草案。 12.提供中药化学对照品的量及其长期计划: (1) 项目完成时,提供纯度达98%以上标准品原则上不少于500mg,纯度达95%以上标准品原则上不少于1000mg; (2) 项目验收后应能继续提供相关 中药化学对照品服务。 五、验收办法及验收标准 在进行中药材质量标准及标准品研究中,为加强项目管理成立中药材质量标准及标准品研究专家组,全程指导并定期检查研究进展,并召开有关研讨会交流经验,以保证课题的顺利进行。 1.专题研究结束后,由科技部生命科学技术发展中心组织专家组统一验收。 2.专题承担单位按技术要求提供完整的书面研究报告,并提供下列资料: (1) 有关中药化学对照品的详细资料及图谱。 (2) 中药材质量标准草案及其起草说明。 3. 按要求将标准品提交科技部生命科学技术发展中心。 4. 经费使用情况报告:国拨经费使用情况及除国家拨款外,地方、部门、承担单位有否配套资金投入;经费使用是否合理。[/size][/td][/tr][/table]
用的是国际方法配制的氨氮标准溶液 不过是按照100ml的比例来配置的 实际取的氯化铵重量误差的范围估计在千分位 (实验室用的万分之一的电子天平) .用的测定方法是HJ535-2009的纳氏试剂分光光度法 但是最近的不知道如何 氨氮曲线都可以达标 相关系数也是3个9以上 斜率都是0.0063(我原来做的曲线斜率范围在0.0074-0.0077) 。具体的出现误差的地方求教是出在哪儿? 所用的水是用的市面上的桶装纯净水(和前面曲线的不是一家的) 酒石酸加纳和纳氏试剂 都是用的现在的水配制的 但是做了几次斜率都是低了 这是为什么
氨氮纳氏试剂法标准曲线斜率有没有相关规定,国标上好像没有,有没有在哪些资料、质控手册上看到过的,斜率在0.0060-0.0070之间靠谱吗?看之前前辈做的好像都在0.0070-0.0080之间。大家有没有看到关于氨氮斜率的相关规定,如果没有的话,你们做的斜率一般在多少?
我们用的GB/T 18204.2-2014,方法是甲醛-酚试剂分光光度法;氨是靛酚蓝分光光度法,请问一下,甲醛、氨标准曲线的斜率具体要求?







 我要推广仪器
我要推广仪器
 下载APP
下载APP