[color=#444444]本人最近按2015版药典做了一个药用辅料-醋酸羟丙甲纤维素琥珀酸酯的的游离乙酸的含量测定实验。实验过程如下:[/color][color=#444444] 游离乙酸、琥珀酸 取本品0.102g,精密称定,置锥形瓶中,精密加入磷酸盐溶液(取0.02mol/L磷酸二氢钾溶液,用1mol/L氢氧化钠溶液调pH值至7.5)4.0ml,搅拌2小时,加磷酸溶液(取1.25mol/L磷酸1ml,置50ml量瓶中,加水稀释至刻度,摇匀)4.0ml,强力振摇,离心,上清液作为供试品溶液;精密称取琥珀酸0.13g,置100ml量瓶中,加水适量,振摇使完全溶解,加水至刻度,摇匀,作为琥珀酸贮备液;取加有水20ml的100ml量瓶,称重,精密加入冰乙酸2ml,再称重,用水稀释至刻度,摇匀,精密量取6ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为乙酸贮备溶液;精密量取乙酸贮备液和琥珀酸贮备液各4.0ml,置同一25ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。照高效液相色谱法(中国药典2015年版四部通则0512)试验。以十八烷基硅烷键合硅胶为填充剂,以0.02moI/L磷酸二氢钾溶液(用6mol/L磷酸溶液调pH值至2.8)为流动相,流速每分钟1ml,检测波长为215nm。取对照溶液10μl, 注入液相色谱仪,按琥珀酸峰计算,理论板数不得少于8000。取供试品溶液与对照溶液各10μl,注入液相色谱仪,按干燥品计算,游离乙酸和琥珀酸总量不得过1.0%。[/color][color=#444444]计算公式: 游离乙酸含量=0.0768(WA/(W(1-干燥失重)))(γUA/γSA)[/color][color=#444444] 式中 WA为乙酸贮备溶液中冰乙酸量,mg;[/color][color=#444444] W为供试品的取样量,mg;[/color][color=#444444] γUA、γSA为供试品溶液、对照溶液中乙酸的峰面积。[/color][color=#444444] 游离琥珀酸含量=1.28(WS/(WUS(1-干燥失重)))(γUS/γSS)[/color][color=#444444] 式中 WS为琥珀酸贮备液中琥珀酸量,mg;[/color][color=#444444] WUS为供试品取样量,mg;[/color][color=#444444] γUS、γSS为供试品溶液、对照溶液琥珀酸的峰面积。[/color][color=#444444]我的问题是,根据“干燥品计算,游离乙酸和琥珀酸总量不得过1.0%”这句话,游离乙酸含量的最后计算的结果要不要乘以100%,比如我最后计算结果是0.0139,如果这个结果再乘以100%,就变为1.39%,从而超过限度,那么就需要重新做实验复核一遍。[/color]
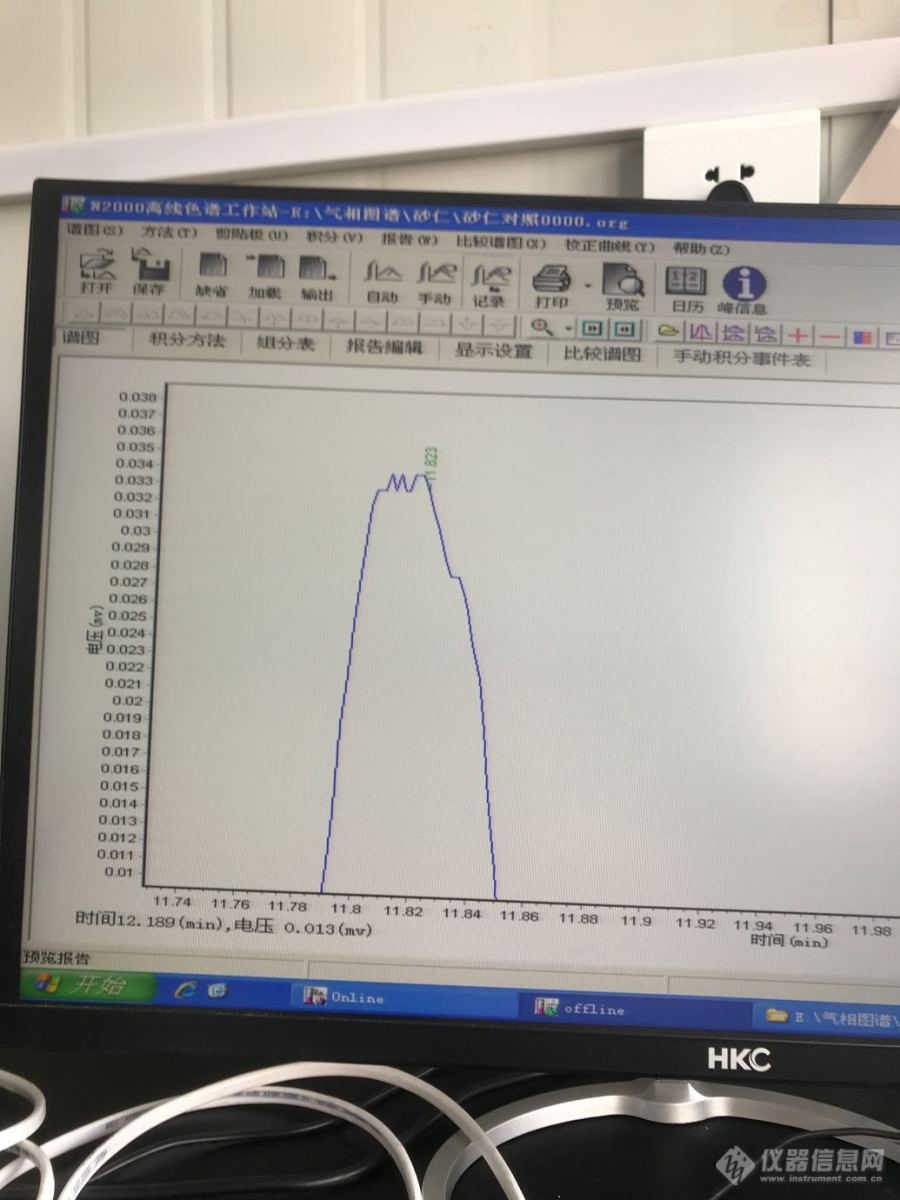
砂仁含量测定,乙酸龙脑脂对照品的峰是这样的,什么原因?[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048233492_354_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048362627_1375_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048546892_2814_4008962_3.png[/img]
今天做乙酸乙酯残留,对照面积分别是702.2,722.5,738.6,790.4,821.2,rsd为6.5%,第一针和最后一针面积相差120左右,但rsd在10%内,这5个对照能采用吗?
我现在在做某物质的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]残留溶剂,标准规定乙酸丁酯≤1000ppm,我配置对照品溶液时可以配置的比标准规定的低吗(比如对照品溶液配成500ppm)?样品中乙酸丁酯的残留大概是80ppm
[color=#333333]求助各位高手帮帮忙,最近在做醋酸纤维素琥珀酸酯乙酸的检测,按照2015版药典方法检测,出峰的时候发现琥珀酸有两个峰出现,不知道是什么原因,琥珀酸的对照买的是麦克林公司的试剂,纯度也都在99.5%。[/color][color=#333333][/color]
目前我们实验室用的维生素A醋酸酯的对照品的供应商断货了,求问一下大家都用的是哪些供应商的对照品?我们也可以去买。我们试用过Sigma的和USP的发现都不行。Sigma的是实际含量和COA上的含量出入较大。USP的是一个混合物有全反式的和CIS的,由于我们不是用的中国药典附录上测定维生素A的方法,所以我们的液相分不开这2种物质,所以也不能用。
我今天按照国标GB/T5750.10-2006中的9测定二氯乙酸,进行的是加标回收。因为是用甲基叔丁基醚做的介质,质谱分析时有很多峰,我无法确定哪一个是二氯乙酸甲酯,自带的质谱库内也没有。请问哪位能帮我找一下二氯乙酸甲酯的质谱分析参数,谢谢。
(1)原理试样经处理后,在pH6左右的溶液中,镉离子与二硫腙形成配合物,并经乙酸丁酯萃取分离,导入原子吸收仪中,原子化以后,吸收228.8nm共振线,其吸收值与镉含量成正比,与标准系列比较定量。(2)试剂 氨水、混合酸、1g/L二硫腙-乙酸丁酯溶液(称取0.1g二硫腙,加10mL三氯甲烷溶解后,再加乙酸丁酯稀释至100rnL,临用时配制)、2mo1/L柠檬酸钠缓冲液(称取226.3g柠檬酸钠及48.46g柠檬酸,加水溶解,必要时加温助溶,冷却后加水稀释至500mL,临用前用1g/L二硫腙-乙酸丁酯溶液处理以降低空白值)、镉标准储备溶液和标准使用液的配制与碘化钾-4-甲基戊酮-2法中的相同。(3)仪器原子吸收分光光度计。(4)分析步骤①试样处理对于谷类要去除其中的杂物及尘土,必要时除去外壳。对于豆类,取可食部分洗净晾干,切碎充分混匀。②样品消化称取5.00g上述试样,置于250mL高型烧杯中,加15mL混合酸,盖上表面皿,放置过夜,再于电热板或电砂浴上加热。消化过程中,注意勿使干涸,必要时可加少量硝酸,直至溶液澄明无色或微带黄色。冷后加25mL水煮沸,除去残余的硝酸至产生大量白烟为止,如此处理两次,放冷。以25mL水分数次将烧杯内容物洗入125mL分液漏斗中。取与处理样品相同量的混合酸、硝酸按同一操作方法做试剂空白试验。③萃取分离 吸取0、0.25mL、0.50mL、1.50mL、2.50mL、3.50mL、5.0mL镉标准使用液(相当于0、0.05μg、0.1μg、0.3μg、0.5μg、0.7μg、1.0μg镉)。分别置于125mL分液漏斗中,各加盐酸(1+11)至25mL。向试样品处理溶液、试剂空白液及镉标准溶液各分液漏斗中各加5mL柠檬酸钠缓冲液(2mol/L),以氨水调节pH至5~6.4,然后各加水至50mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),以氨水调节pH至5~6.4,然后各加水至501mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),振摇2min,静置分层,弃去下层水相,将有机层放入具塞试管中,备用。④测定测定方法与碘化钾-4-甲基戊酮-2法中的相同。⑤结果计算 样品中镉的含量按下式进行计算。X=/(m×1000)式中,X为试样中镉的含量,mg/kg;A1为测定用试样液中镉的质量,μg;A2为试剂空白液中镉的质量,μg;m为试样质量或体积,g或mL。计算结果保留两位有效数字。⑥精密度 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%。
我实验要在负离子模式下定量测定一种代谢物,但买到的对照品是三氟乙酸盐结合的形式,在论坛里看到说,三氟乙酸不能再负离子模式下使用,会抑制响应,那我的对照品就不能用了吗?应该怎么办呢?
有谁能告诉我维生素A乙酸酯标准品99%的都买的哪里的?
[align=right][b]SGLC-GC-039[/b][/align][b]摘要:[/b]本文建立了砂仁中乙酸龙脑酯含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件并对升温程序进行优化,采用色谱柱SH-1 分析砂仁中乙酸龙脑酯,乙酸龙脑酯峰形对称,理论塔板数按乙酸龙脑酯峰计算远高于10000,满足《中国药典》要求。此方法可为砂仁中乙酸龙脑酯含量测定提供参考。。[b]关键词:[/b]砂仁 乙酸龙脑酯 SH-1 GC[b]1. 实验部分1.1 实验仪器及耗材[/b]Shimadzu GC-2030[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url];色谱柱:SH-50(30 m,0.25 mm × 0.25 μm;P/N:227-36162-01;S/N:1553669);SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05);[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34002-01);SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02);SHIMSEN Pipet PMII-100(P/N:380-00751-04);SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。[b]1.2 对照品溶液的制备[/b]取芳樟醇对照品适量,精密称定,加乙酸乙酯制成每1 mL含0.1 mg的溶液,即得。[b]1.3 供试品溶液的制备[/b]取本品粉末(过三号筛)约1 g,精密称定,置具塞锥形瓶中,精密加入无水乙醇25 mL,密塞,称定重量,超声处理(功率300W,频率40 kHz)30 分钟,放冷,用无水乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。。[b]1.4 分析条件[/b]色谱柱:SH-1 (30 m, 0.25 mm × 0.25 μm P/N:221-75719-30;S/N:1541069 )升温程序:初始温度80 ℃,保持1分钟,以每分钟2 ℃的速率升温至100 ℃,保持5分钟载气:N2进样口温度:230 ℃分流模式:分流(10:1)控制模式:恒线速度(30 cm/s)初始流速:1.09 mL/min检测器:FID,温度:250 ℃进样量:1 μL[b]2. 实验结果[/b]按照上述色谱条件(1.4)进行采集,对照品溶液和供试品溶液色谱图如下:[b]对照品溶液[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_1.png[/img][font=arial, &][size=12px][/size][/font][b]供试品溶液[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_2.png[/img][font=arial, &][size=12px][/size][/font][b]重现性[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_3.png[/img][font=arial, &][size=12px][/size][/font][b]3. 结论[/b]本文建立了砂仁中乙酸龙脑酯含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件,采用色谱柱SH-1 分析砂仁中乙酸龙脑酯,乙酸龙脑酯峰形对称,理论塔板数按乙酸龙脑酯峰计算远高于10000,满足《中国药典》要求。此方法可为砂仁中乙酸龙脑酯含量测定提供参考。
[b][font=宋体]问题描述:[/font][font=宋体]按照国标[/font]GB5009.121-2016[font=宋体]方法,以[/font]0.02mol/L[font=宋体]乙酸铵:甲醇([/font][i]V/V[/i][font=宋体])[/font]=90+10[font=宋体],流速[/font]1mL/min[font=宋体],可以检测脱氢乙酸,但在流动相中加入[/font]0.1%[font=宋体]的乙酸后,同样的分析时间内却检测不到脱氢乙酸色谱峰,这是为什么?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])脱氢乙酸是我国允许在食品生产加工中使用的一种化学合成防腐剂,在较高的[/font]pH[font=宋体]范围内保持良好的抗菌性。脱氢乙酸是极性化合物,因其含有双键具有紫外吸收,在[/font]C[sub]18[/sub][font=宋体]色谱柱有一定的吸附保留。在[/font]GB 5009.121-2016[font=宋体]《食品安全国家标准[/font] [font=宋体]食品中脱氢乙酸的测定》[/font] [font=宋体]液相色谱法中使用[/font]0.02mol/L[font=宋体]乙酸铵:甲醇([/font][i]V/V[/i][font=宋体])[/font]=90[font=宋体]:[/font]10[font=宋体]作为流动相,对脱氢乙酸作等度洗脱。[/font][font=宋体]([/font]2[font=宋体])通常可电离化合物而言,不同状的化合物态与[/font]C[sub]18[/sub][font=宋体]之间的作用力存在明显差异。脱氢乙酸[/font]p[i]Ka[/i][font=宋体]值为[/font]5.27[font=宋体],而[/font]0.02mol/L[font=宋体]乙酸铵:甲醇([/font][i]V/V[/i][font=宋体])[/font]=90[font=宋体]:[/font]10[font=宋体]流动相体系的[/font]pH[font=宋体]值在[/font]7~8[font=宋体],在此体系中,大部分脱氢乙酸处于离子态。当原有体系中加入乙酸酸化后,流动相体系的[/font]pH[font=宋体]值有利于脱氢乙酸从离子态向中性态转化,从而促使脱氢乙酸与[/font]C[sub]18[/sub][font=宋体]的作用力更强了,因此,相对而言,乙酸铵:甲醇([/font][i]V/V[/i][font=宋体])[/font]=90[font=宋体]:[/font]10[font=宋体]流动相洗脱能力变弱了,导致脱氢乙酸在[/font]C[sub]18[/sub][font=宋体]色谱柱上的保留时间更长,或长期保留在色谱柱上。只有延长酸化后流动相的洗脱时间,或增加酸化后流动相中有机相的比例,可将脱氢乙酸从色谱柱中洗脱出来。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]
如何检测石油醚,乙酸甲酯和丙酮???石油醚本身就有几个峰,要做内标法找不到对照品。用异丙醇或正丙醇做内标物可以嘛?哪个好一些?DB-WAX柱子?进样口、柱温、检测器温度如何??谢谢
[color=#333333]求助各位高手帮帮忙,最近在做醋酸纤维素琥珀酸酯乙酸的检测,按照[/color][color=#333333]2015[/color][color=#333333]版药典方法检测,出峰的时候发现琥珀酸有两个峰出现,不知道是什么原因,琥珀酸的对照买的是麦克林公司的试剂,纯度也都在[/color][color=#333333]99.5%[/color]
大家经常做气相需要测对照品溶液,有时对照品溶液的出峰面积在不同时期可能差异较大,大家遇到过这样的问题吗?问题:取甲醇、乙酸乙酯、甲苯适量,精密称定,用DMF溶解稀释定容制成每1ml中约含甲醇60ug、乙酸乙酯100ug、甲苯20ug的混合溶液。那么我们实际称取的对照质量应该在什么范围内是可以接受的?因为称量的差异会导致对照品出峰面积的差异。问题:如何能用天平称准对照试剂的量?(有机溶剂甲醇、乙腈、二氯甲烷等)换算成体积量取还是直接称取?
一个香草香精,里面有乙酸和香兰素,在后面发现了乙酸香兰素酯,请问这个是添加了,还是缩合而成的?谢谢
[font=SimSun, STSong, &]我的食品生产许可证上的执行标准是SB/T10439-2007产品分类是酱腌菜产品明细是盐水食用菌,请问的我生产的盐水食用菌允许使用乙二胺四乙酸二钠吗?2760上边按照酱腌菜的标准是允许的,按食用菌制品的标准是不允许使用的,我是应该按照食用菌制品的标准不允许使用呢还是应该按照生产许可证上的酱腌菜标准允许使用呢?有没有专业人士帮忙解答啊?[/font]
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font][/b][font=宋体][/font][b][font=宋体][/font][/b] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font][font=宋体] [/font] [font=宋体][/font] [font=宋体](1)本品粉末棕黄色。管胞常成束,多断裂,直径10~81μm,圆形[color=var(--weui-LINK)]具缘纹孔[i][/i][/color]明显;具缘纹孔单列于管胞壁,直径近等于管胞直径。射线管胞壁锯齿状增厚,交叉场纹孔窗格状。树脂团块不规则,棕黄色或棕红色。[/font] [font=宋体](2)取〔含量测定〕项下的挥发油0.1ml,加乙酸乙酯1ml使溶解,作为供试品溶液。另取[color=var(--weui-LINK)]α[/color][color=var(--weui-LINK)]-[/color][color=var(--weui-LINK)]松油醇[i][/i][/color]对照品,加乙酸乙酯制成每1ml含10μl的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各1μl,分别点于同一硅胶G薄层板上,以[color=var(--weui-LINK)]石油醚[i][/i][/color](30~60℃)-乙酸乙酯(17:3)为展开剂,展开,取出,晾干,喷以香草醛硫酸试液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]测定[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][b][font=宋体][/font][/b] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] [b]挥发油[/b] 照挥发油测定法(通则2204甲法)测定。[/font] [font=宋体]本品含挥发油不得少于0.40%(ml/g)。[/font] [b][font=宋体]α-蒎烯[/font][/b][font=宋体] [/font][font=宋体]照气相色谱法(通则0521)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]弹性石英毛细管柱(柱长为30m,内径为0.32mm,膜厚度为0.25μm)DB-5(交联5%苯基甲基聚硅氧烷为固定相);程序升温;初始温度60℃,保持5分钟,以每分钟5℃的速率升温至160℃,然后以每分钟70℃的速率升温至300℃,保持10分钟;进样口温度为200℃;检测器温度为320℃;[color=var(--weui-LINK)]分流比[i][/i][/color]为5:1。理论板数按α-蒎烯峰计算应不低于25 000。[/font] [b][font=宋体]对照品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取α-蒎烯对照品适量,精密称定,加乙醇制成每1ml含0.2mg的溶液,即得。[/font] [b][font=宋体]供试品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取本品粉末(过三号筛)约2g,精密称定,置具塞锥形瓶中,精密加入乙醇20ml,密塞,称定重量,超声处理(功率150W,频率50kHz,水温30℃以下)15分钟,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。[/font] [b][font=宋体]测定法 [/font][/b][font=宋体]分别精密吸取对照品溶液与供试品溶液各1μl,注入气相色谱仪,测定,即得。[/font] [font=宋体]本品按干燥品计算,含α-蒎烯(C[sub]10[/sub]H[sub]16[/sub])不得少于0.10%。[/font] [font=宋体][/font]
请问哪位老师有做水中二氯乙酸和三氯乙酸含量的,前处理的液液萃取和衍生怎样做比较方便,有没有什么设备可以轻松解决啊?谢谢!

前几天同事扩项工作场所空气中饱和脂肪酸酯类物质包括乙酸甲酯,乙酸乙酯,乙酸丙酯,乙酸丁酯,标准是GBZ/T160.63-2007。柱子是SH-Rtx5(30m*0.32mm*0.25um),同事欲恒温同时分离这四种酯类,我提示乙酸乙酯,乙酸丙酯,乙酸丁酯混一起恒温做没事,如果乙酸甲酯也混一起做那么会与溶剂二硫化碳峰难分离,于是他计划先做乙酸乙酯,乙酸丙酯,乙酸丁酯再另外单独做乙酸甲酯。 乙酸乙酯,乙酸丙酯,乙酸丁酯色谱条件:岛津气相色谱GC2010PLUS 柱温60℃ 检测器进样器均为200℃ 分流比50 恒线速度22cm/shttp://ng1.17img.cn/bbsfiles/images/2017/01/201701191702_673938_2103464_3.jpg三种乙酸酯峰型还不错。接下来他说想试试乙酸甲酯在这个条件下出峰会怎么样?因为我之前用OV101做过二硫化碳中乙酸甲酯,它是紧挨着在二硫化碳前出峰,同时也用DB-FFAP做过它是在二硫化碳之后。SH-Rtx5极性比OV101强些 比DB-FFAP弱很多,那么在二硫化碳之前还是二硫化碳之后出峰呢? 看到甲乙丙丁突然有了一想法:不是有碳数规律吗?利用碳数规律推测乙酸甲酯的保留时间:保留时间:乙酸乙酯 2.854min 乙酸丙酯 3.562min 乙酸丁酯5.162min 二硫化碳2.612min碳数规律:lnt‘=An+C t’为调整保留时间 n为同系物中碳个数 A, C均为常数首先精确计算死时间:间隔均匀同系物 精确死时间计算公式:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616020890_01_2103464_3.pngtm=(2.854*5.162-3.562*3.562)/2.854+5.162-3.562-3.562=2.292min调整保留时间代入碳数规律公式:乙酸乙酯ln0.562=4A+C 乙酸丙酯 ln1.27=5A+C 求得乙酸甲酯调整保留时间lnt’=3A+C t’=0.249min乙酸甲酯的预测保留时间0.249+2.292=2.541min.这个保留时间在二硫化碳(2.612min)之前,两者仅仅相差0.07min。于是预测同条件下乙酸甲酯在二硫化碳之前出峰并且分离度不好!同条件实验做二硫化碳中乙酸甲酯3000ug/ml来验证:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616132954_01_2103464_3.jpg实验结果乙酸甲酯保留时间是2.571min与预测的2.541min比较符合,外推是有误差的,而且本例碳数不多,碳数多些会更准确。降低柱温至40℃,线速度15cm/s 乙酸甲酯与二硫化碳分离达到定量要求!http://ng1.17img.cn/bbsfiles/images/2016/09/201609061616_608604_2103464_3.jpg 结论:碳数规律还是比较准的,可以预测出峰情况。
各位老师,小弟最近在实验室中用气质(瓦里安3800)测香精样品,但是在分析乙酸乙酯时发现找不到对应峰。于是跑了一针乙酸乙酯标样(浓度30ppm,溶剂甲基叔丁基醚),但是还是找不到对应峰,请各个专家指点一二,看看问题究竟出在哪儿。谱库中乙酸乙酯的图谱及标品分析后的谱图如下,请参考。乙酸乙酯的标准质谱图:http://ng1.17img.cn/bbsfiles/images/2017/01/201701231132_01_2297325_3.bmphttp://ng1.17img.cn/bbsfiles/images/2017/01/201701231132_01_2297325_3.bmp乙酸乙酯标样图谱http://ng1.17img.cn/bbsfiles/images/2017/01/201701231134_01_2297325_3.bmp
乙酸对叔丁基环己酯 和乙酸邻叔丁基环己酯如何区分?谢谢
请问有谁做关于食品接触塑料中总迁移量的检测和乙酸乙烯酯的检测?标准上说水模拟液加丙酮定容后,就可以上GC-MS上检测,但是丙酮中含有水,这样的操作合适么?对仪器,对色谱柱会不会有损害?
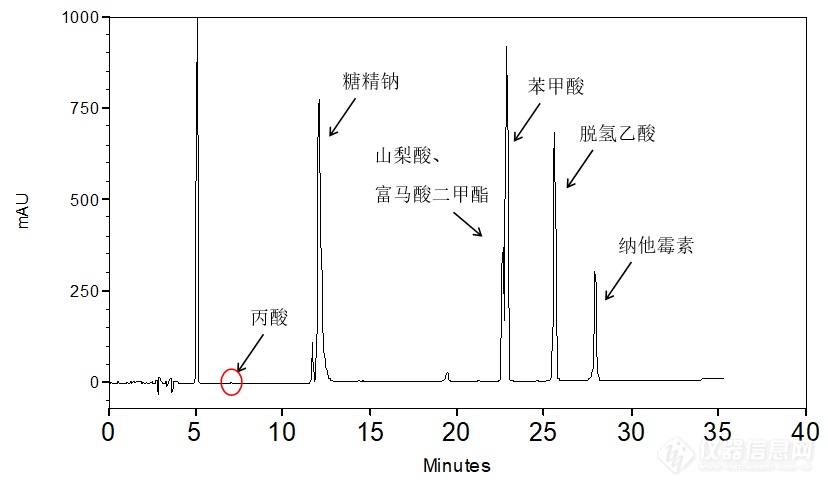
[align=center][b]食品中7种添加剂的共同分析——丙酸钠、山梨酸、脱氢乙酸、苯甲酸、糖精钠、纳他霉素和富马酸二甲酯[/b][/align][img=,99,45]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143105100_953_2222981_3.gif!w99x45.jpg[/img] [img=,121,46]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143267296_3413_2222981_3.gif!w121x46.jpg[/img] [img=,98,83]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143361658_9073_2222981_3.gif!w98x83.jpg[/img] [img=,89,50]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143485158_3959_2222981_3.gif!w89x50.jpg[/img] 丙酸钠 山梨酸 脱氢乙酸 苯甲酸[img=,112,150]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144042246_934_2222981_3.gif!w112x150.jpg[/img] [img=,270,166]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144144504_7297_2222981_3.gif!w270x166.jpg[/img] [img=,123,58]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144263678_2065_2222981_3.gif!w123x58.jpg[/img] 糖精钠 纳他霉素 富马酸二甲酯丙酸钠、山梨酸、脱氢乙酸、苯甲酸、糖精钠、纳他霉素和富马酸二甲酯为食品中常见的添加剂分析项目,客户希望通过一种方法来同时实现7种添加剂的分析,且整体分析时间要求尽量短,以提高工作效率。首先,使用大曹三耀(原资生堂)CAPCELL PAK系列色谱柱中的第一选择色谱柱——通用型反相柱[color=#3366ff][b]CAPCELL PAK C18 MGII[/b][/color]来进行方法初筛;考虑到7种添加剂中的酸性化合物较多,为取得良好保留,在流动相的选择方面,首先尝试在酸性条件下(0.1%磷酸)进行分析,结果如图1所示。在酸性条件下,苯甲酸、山梨酸和富马酸二甲酯三者间分离情况不佳。[align=center][img=,690,405]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104146524_289_2222981_3.png!w690x405.jpg[/img][/align][align=center]图1 酸性条件下分析结果[/align][img=,431,219]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104139552_9327_2222981_3.png!w431x219.jpg[/img]在此条件下,进一步对梯度条件进行调整,并将流动相中的乙腈更换为甲醇,更换不同种类色谱柱,但均未能获得良好结果。接下来我们将流动相pH改变,将0.1%磷酸溶液更换为20 mmol/L磷酸二氢铵溶液进行分析,结果如图2所示。能够看到7个较明显物质峰得到分离。[align=center][img=,690,359]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104574536_4474_2222981_3.png!w690x359.jpg[/img][/align][align=center]图2 磷酸二氢铵条件分析结果[/align][img=,450,220]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104576746_74_2222981_3.png!w450x220.jpg[/img]由于客户想要在短时间内得到良好分析,因此我们进一步将色谱柱由常规规格[b]S5 4.6 mm i.d. × 250 mm[/b]更换为[b][color=#3366ff]S3 4.6 mm i.d. × 100 mm[/color][/b],分析结果如图3所示。[align=center][img=,690,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191105363445_4814_2222981_3.png!w690x418.jpg[/img][/align][align=center]图3 MGII小粒径短柱分析结果[/align][img=,459,221]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191105376737_3055_2222981_3.png!w459x221.jpg[/img]但同时在分析中发现丙酸钠单标无法得到良好响应(图4),且富马酸二甲酯出峰行为异常(图5)。[align=center][img=,690,337]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191106365666_7879_2222981_3.png!w690x337.jpg[/img][/align][align=center]图4 丙酸钠单标分析结果[/align][align=center] [/align]如图5结果所示,将7种添加剂各取100 μL后稀释到1 mL配制混合标准品时(第一次混合样品,终浓度为各0.1 mg/mL),未见富马酸二甲酯峰出现,此时再将此溶液与富马酸二甲酯单标以4:1比例混合后(第二次混合样品,富马酸二甲酯终浓度0.28 mg/mL,其他为0.08 mg/mL),富马酸二甲酯峰出现,同时2号峰峰面积增大。富马酸二甲酯单标进样无异常。因此富马酸二甲酯在混合溶液中可能存在[color=red]降解反应、溶解度不佳或其它原因[/color],导致出峰行为异常,建议对该物质性质进行考察,或进行方法学验证。[align=center][img=,690,450]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191106370947_4629_2222981_3.png!w690x450.jpg[/img][/align][align=center]图5 富马酸二甲酯分析结果[/align][align=left] 综上所述,使用[b][color=#ff0000]CAPCELL PAK C[sub]18[/sub] MGII S3 4.6 mmi.d. × 100 mm[/color][/b]色谱柱可在[color=#3366ff][b]20 mmol/L磷酸二氢铵-乙腈[/b][/color]条件下基本实现7种常见食品添加剂的分析,但丙酸和富马酸二甲酯出峰异常,建议进一步进行方法学验证。[/align][align=left][/align][align=left] [/align][align=right] [/align][b][/b][align=right][b][color=#333333]LC Application Lab, Sanyofine China[/color][/b][/align][color=#333333][/color][align=right]Beijing, China[/align][color=#333333][/color][align=right]Phone 400 801 3103[/align]
据广东出入境检验检疫机构称,在日本进口的日式酱油、芥末酱中检测出了甲苯和乙酸乙酯。有关食品产自三家日本生产企业。其中甲苯的最高检出值为0.0053mg/kg,乙酸乙酯最高检出值为0.537mg/kg。此前,有日本媒体报道,日本有人食用了检出甲苯和乙酸乙酯的食品出现过不适症状。中国国家质检总局称,进口上述产品的中国进口企业,已开始对这三家企业生产的同类产品采取下架和批批检验措施,以确保中国消费者安全。甲苯 健康危害:对皮肤、粘膜有刺激性,对中枢神经系统有麻醉作用。 慢性中毒:长期接触可发生神经衰弱综合征,肝肿大,女工月经异常等。皮肤干燥、皲裂、皮炎。乙酸乙酯用作清漆稀释剂,人造革、硝酸纤维素塑料等的溶剂,也用作染料、药物、香精等的原料。作为增香剂用于食品和饲料中。
离子色谱法测定水中二氯乙酸和三氯乙酸检测实施细则1 目的规范离子色谱测定生活饮用水和水源水中二氯乙酸和三氯乙酸检测方法,保证检测工作顺利进行。2 方法的适用范围适用于使用戴安ICS-900离子色谱仪对生活饮用水和水源水中二氯乙酸和三氯乙酸进行检测。该方法对这两种消毒副产物的方法检出限(MDL)分别为:二氯乙酸:1.5μg/L;三氯乙酸:1.8μg/L,定量检测限(LOQ)分别为:二氯乙酸:6.0μg/L三氯乙酸:7.2μg/L。3 仪器3.1 ICS-900离子色谱仪:AS19分析柱、AG19保护柱、ASRS300抑制器、ED50电导检测器3.2 AS-DV自动进样器3.3 CR-ATC淋洗液自动生成器4 试剂4.1 纯水:含各种待测阴离子应低于仪器的最低检出限,使用前应测定空白值。4.2 二氯乙酸和三氯乙酸标准物质,由上海安谱科学仪器有限公司提供。二氯乙酸标准品:1g;三氯乙酸标准品:1g(纯度为99.40%)。5 水样5.1 水样的采集及储存:水样采集在聚乙烯瓶中,于4℃冰箱内保存。5.2 水样的预处理:经0.2μm孔径的微孔滤膜过滤后可直接进样;若样品中碳酸根的质量浓度在200 mg/L以上,则要经过酸化除去大量存在的碳酸根,然后将样品在真空装置中进行脱气,再进样分析。样品测定时应注意的问题样品中某些组分浓度太高,应选择适当的稀释倍数稀释样品后测定。如果已检测了高组分的样品,并对下一样品的测定产生了影响,建议进1一2针去离子水测定,将高组份完全洗脱后再测下一个样品。由于进样量小,操作装应严格防止纯水和器皿在水样预处理过程中的污染,以确保分析的准确性。6 试验6.1 色谱条件进样体积:300μL;流量:1.0mL/min。表5.1.1.1 淋洗液梯度程序时间/(min)氢氧化钾浓度/(mmol/L)0.010.025.0
请问哪里可以买到敌鼠钠盐标准品和氟乙酸标准品啊?
请大家看看,一般乙酸二氢香芹酯是几个峰,我这里认为是后面两个大峰,但不知道前面的一个9.592的峰是什么,大家看看。
各位老师们好,我正在用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]测定乙酸残留,检测器FID,柱子为DB-FFAP,进样口150,检测器240,进样量1ul,流速3.4ml/min,分流比1:1,程序升温为50度,保持3min,以30度每分钟升温到200度,保持5min,稀释液为甲醇,前面刚开始测对照时,乙酸正常出峰,后面一测样品,乙酸就没出峰,加标供试品中乙酸也没出峰,且进完供试品后再进对照,对照中的乙酸也不出峰了,但是溶剂峰还能正常出峰,不知道是不是样品对柱子污染了导致乙酸不出峰[img=,690,169]https://ng1.17img.cn/bbsfiles/images/2023/10/202310111422246766_4558_5416762_3.png[/img][img=,690,140]https://ng1.17img.cn/bbsfiles/images/2023/10/202310111422250502_9836_5416762_3.png[/img]
[align=center][font='方正小标宋简体'][size=29px]巧用乙酸除铜绿[/size][/font][/align][font='仿宋_gb2312'][size=21px]我们实验室的赛默飞的[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICP-MS[/color][/url]购于2016年,已经使用7年,我来到实验室的时候发现上面已经长了不少绿色的物质,老师们说这个不影响实验结果,但是我想让事情是它本来该有的那个样子,所以我想试着把它清洗干净。[/size][/font][font='仿宋_gb2312'][size=21px]第一步,我分析这个绿色的物质可能是铜绿,因为我觉得这个大块金属是铜制的,查询了仪器说明书,确定是铜制的金属件后,我大胆认为是碱式碳酸铜,化学式为Cu[/size][/font][font='仿宋_gb2312'][sub][size=21px]2[/size][/sub][/font][font='仿宋_gb2312'][size=21px](OH)[/size][/font][font='仿宋_gb2312'][sub][size=21px]2[/size][/sub][/font][font='仿宋_gb2312'][size=21px]CO[/size][/font][font='仿宋_gb2312'][sub][size=21px]3[/size][/sub][/font][font='仿宋_gb2312'][size=21px]。[/size][/font][font='仿宋_gb2312'][size=21px]第二步,确定了是铜绿后,在网上查询了如何去除铜绿,有的说用乙酸和食盐;有的说用柠檬;有的说用白醋和面粉;有的说用铜清洁剂。考虑实验室已有的试剂,我打算使用纯乙酸进行清洗。首先,乙酸的酸性比碳酸强,可以与铜绿反应;其次,乙酸酸性弱,不与铜单质反应,不会腐蚀器件。[/size][/font][font='仿宋_gb2312'][size=21px]第三步,准备好乙酸、去离子水、棉签、擦镜纸。用棉签充分浸泡乙酸先简单的把有铜绿的部分全部涂抹一遍(乙酸的刺激性较强,需要戴好口罩),等一分钟,就可以开始用力擦,最不容易清洗的位置是那两道沟,用剪刀把擦镜纸剪成小块,对折两次,把回形针掰直,顶着蘸有乙酸的擦镜纸来回擦拭,铜绿完全去除后,就用去离子水润湿整张擦镜纸,反复擦拭乙酸涂抹过的区域,重复五次保证乙酸完全清除,整个过程用时90分钟左右,下面是对比图。[/size][/font][img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309010940432703_8417_5814833_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309010940433780_4938_5814833_3.jpeg[/img][font='仿宋_gb2312'][size=21px]这件事让我更加理解什么是知易行难,从我看到铜绿到开始行动去除铜绿,大约有两个月的时间,从我动手去除铜绿到清洗完毕,只用了不到两个小时的时间,知道与做到有一条巨大的鸿沟,回首工作中的点点滴滴,一道道的沟也记不清是何时走过,只是未来的路上仍不平坦,让我们身披坚强的盔甲,跨上勇气的战马,手执行动的利剑,前进四,出发![/size][/font]







 我要推广仪器
我要推广仪器
 下载APP
下载APP