高效液相色普法测定甘草酸苷含量,甘草酸苷对照品峰面积与之前相比较偏高,导致含量偏高,请问有可能是哪些原因?
http://ng1.17img.cn/bbsfiles/images/2012/09/201209272130_393425_2255248_3.gifHPLC测定木犀草苷对照品为什么峰前面有个小峰????
[b]Q:四君子颗粒中甘草苷、甘草酸铵的检测,供试品溶液的前处理步骤是?A:供试品溶液:取本品装量差异项下的内容物3g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,密塞,称定重量,超声处理(功率250W,频率40KHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液15ml,蒸干,残渣加甲醇使溶解,移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。===============================================================【活动内容】1、每个工作日上午10:00左右发布一个关于应用数据库的应用问答题,版友根据题目给出自己理解的答案。2、每个工作日下午15:10公布参考答案。【活动奖励】幸运奖:抽奖软件,当天随机抽取3个或5个回答正确的版友ID号(最后一个ID号,截止至下午15:00),每人奖励[color=#ff0000]2钻石币[/color](抽奖人数≤10,抽取3个版友;抽奖人数>10,抽取5个版友);莫名其妙(注册ID:moyueqiu)999youran(注册ID:999youran)yy_0324(注册ID:yy_0324)大川之子,纵横四海(注册ID:chuangu120)dahua1981(注册ID:dahua1981)[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2018/12/201812171515416757_7540_1610895_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2018/12/201812171516149100_7965_1610895_3.png!w690x387.jpg[/img]积分奖励:所有回答正确的版友奖励[color=#ff0000]10个积分[/color](幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次[/b][align=left][color=#ff0000][b]PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。[/b][/color][/align][align=left][color=#ff0000][b] 下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。[/b][/color][/align][align=center]=======================================================================[/align]方法:HPLC基质:药品应用编号:103534化合物:甘草苷、甘草酸铵色谱柱:[url=http://www.dikma.com.cn/product/details-855.html]Platisil ODS 5μm 250 x 4.6mm[/url]样品前处理:1、对照品溶液:取甘草苷对照品、甘草酸铵对照品适量,精密称定,加甲醇制成每1ml分别含甘草苷20μg、甘草酸铵0.2mg溶液,即得(甘草酸重量=甘草酸铵重量/1.0207)。2、供试品溶液:取本品装量差异项下的内容物3g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,密塞,称定重量,超声处理(功率250W,频率40KHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液15ml,蒸干,残渣加甲醇使溶解,移至5ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。色谱条件:色谱柱: Platisil ODS 250*4.6 mm,5 μm(Cat#:99503)流动相: A:乙腈 B:0.05%磷酸溶液流速: 1.0 mL/min柱温: 30 ℃检测器: UV 237 nm进样量: 10 μL文章出处:天津应用实验室关键字:四君子颗粒、甘草苷、甘草酸铵、Platisil C18、HPLC、2015药典摘要:Platisil C18检测四君子颗粒中甘草苷、甘草酸铵。图谱:[img]http://www.dikma.com.cn/u/image/2015/08/04/1438671548996616.png[/img][img]http://www.dikma.com.cn/u/image/2015/08/04/1438671551486259.png[/img][img]http://www.dikma.com.cn/u/image/2015/08/04/1438671554462333.png[/img]

问题:四君子颗粒中甘草苷、甘草酸铵的检测对照品分析中甘草苷与甘草酸铵的分离度是?答案:62.445【活动奖励】因zgx3025(注册ID:v2844608)的答案不正确,所以取消本次获得的钻石币幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币mengzhaocheng(注册ID:mengzhaocheng)莫名其妙(注册ID:moyueqiu)http://ng1.17img.cn/bbsfiles/images/2016/03/201603031621_585902_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603031621_585903_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================四君子颗粒中甘草苷、甘草酸铵的检测样品制备制备方法1. 对照品:取甘草苷对照品、甘草酸铵对照品适量,精密称定,加甲醇制成每1 mL分别含甘草苷20 μg、甘草酸铵0.2 mg溶液,即得(甘草酸重量=甘草酸铵重量/1.0207)。2. 供试品:取本品装量差异项下的内容物3 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定重量,超声处理(功率250 W,频率40 KHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液15 mL,蒸干,残渣加甲醇使溶解,移至5 mL量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。分析条件色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相A:乙腈 B:0.05%磷酸溶液 梯度流速1.0 mL/min柱温30 ℃检测器UV 237 nm 进样量10 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/03/201603031020_585805_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 15.739 771814 49202 22131.352 0.998 -- 2 36.170 766340 93054 391608.534 1.043 62.445 *药典要求理论板数按甘草苷峰计算应不低于5000供试品http://ng1.17img.cn/bbsfiles/images/2016/03/201603031021_585807_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 15.784 475765 27766 18773.718 0.973 -- 2 36.033 152478 18510 403100.536 0.997 58.879 *药典要求理论板数按甘草苷峰计算应不低于5000本品种同时使用了Diamonsil C18、DiamonsilC18(2)两款色谱柱,在药典规定条件下进行甘草苷、甘草酸铵的检测,均满足药典要求。
对照品甘草苷 后面出杂峰 是怎么回事[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221432219383_6429_3461163_3.jpeg[/img]
最近做小儿七星茶颗粒甘草酸的测定时遇到了很奇怪的问题:对照品有峰,样品却只出杂质峰,开始怀疑是样品没有含量,可是拿以前做过的有含量的样品再做,却没峰了;后来吸一半对照品一半样品进样就出峰了,可是加对照品到样品中一起按标准处理后就又没有峰出来,怎么想都不明白问题出在哪里。所用的试剂换了好几次,也换人配了,结果还是一样没有,会不会是超声引起的呢,因为我们的超声机的功率只有80瓦,不过以前也做得出啊,大家帮帮忙,下面是标准【含量测定】 照高效液相色谱法(中国药典2005年版一部附录VI D)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.2mol/L醋酸铵溶液—冰醋酸(65:35:1)为流动相;检测波长为250nm。理论板数按甘草酸峰计算应不低于2000。 对照品溶液的制备 取甘草酸铵对照品适量,精密称定,加流动相制成每1ml含16μg的溶液,即得(折合甘草酸为15.672μg)。 供试品溶液的制备 取装量差异项下的本品内容物,混匀,研细,取约7g,精密称定,置50ml量瓶中,加流动相约45ml,超声处理(功率300W,频率40kHz)30分钟,放冷,加流动相至刻度,摇匀,滤过,取续滤液,即得。 测定法 精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得。
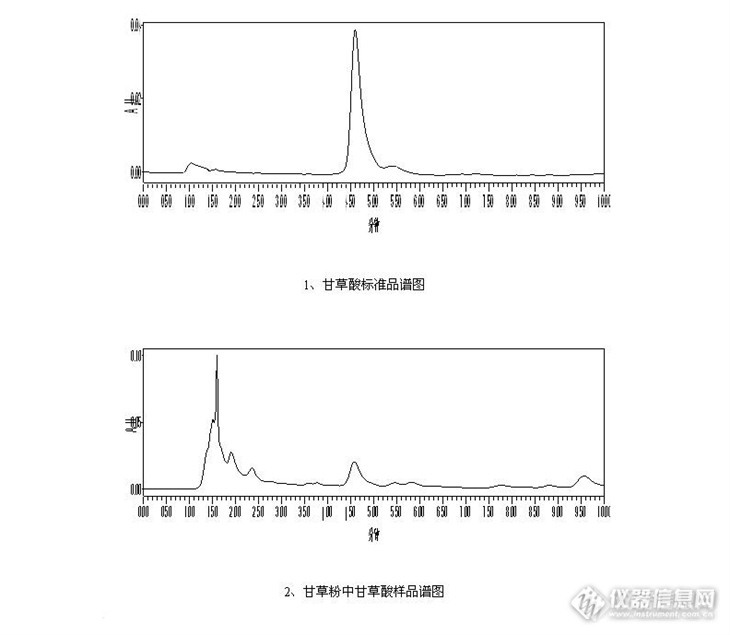
高效液相色谱法测定甘草制品中甘草酸的含量前言甘草酸是一味常用的中药,目前有关甘草的研究很多,甘草具有清热解毒,润肺止咳之功效。同时,我们也要注意甘草的副作用,过量的甘草会使到尿量及钠的排出减少,身体会积存过量的钠(盐分)引起高血压;水分储存量增加,会导致水肿。同时过多血钾流失引起的低血钾症,导致心律失常,肌肉无力。所以有高血压症状的人是不能食用甘草的,我们的父母有一种误区,觉得食用甘草可以清热解毒,殊不知其副作用也同样可怕。中药无毒论确实非常可怕啊!闲话少说,回到正题。http://ng1.17img.cn/bbsfiles/images/2013/09/201309190950_465407_2428063_3.jpg甘草切片的图片甘草的主要成分为甘草酸、甘草次酸和甘草苷等。甘草酸是最主要的有效成分。目前,我们使用的检测方法为高效液相色谱法,同时又该方法又有等度高效液相法和梯度高效液相色谱法。1、如果单纯检测甘草酸,我建议使用普通的高效液相色谱法,本单位所使用色谱柱为:Sunfire TM C18色谱柱,填料为5um,规格为4.6*150mm。所使用的仪器为Waters 2695,配备了2887 PDA全波段紫外检测器,可以根据需要提取不同波长的色谱图。如下为:甘草酸的色谱分离条件甲醇-0.2mol醋酸铵-冰醋酸(66:33:1) 检测波长为250nm 流速为1mL/min样品提取过程称取0.2克样品,加入1:1的甲醇水溶液,超声波提取20分钟,然后定容到100mL的容量瓶中。http://ng1.17img.cn/bbsfiles/images/2013/09/201309190952_465408_2428063_3.jpg2、本人查询了最新的2010版药典,发现检测的方法发生了变化,其核心变化就是检测的物质多了一个甘草苷,可以同时实现两种物质的分离,从分离学上来讲是非常有意义的。但是如果单纯的测定甘草酸,我觉得还是使用2005版药典的方法更好。以前的方法5分钟左右就可以出峰,10分钟就可以完成检测。但是采用新方法以后,一针样品要运行1个小时,对色谱柱和时间都是极大的浪费,当然在检测过程中,我们可以进行适当的优化,但是无疑是很劳民伤财的。
大家都用那些容积配置过黄芩苷对照品啊?怎么我们用稀乙醇溶液溶解的时候不易溶解啊,超声半小时还会有很多不容物?
甘草提取液,同样的条件,在Agilent ZOBAX Eclipse plus C18 和迪马铂金C18上跑出来的图相比,前者就硬是差一个最大的峰----甘草酸,用对照品进样也发现在前者上不出峰,后用100%乙腈冲很久才出来一大堆杂质峰,应该是吸附在柱子上了。请问这2种都是C18的柱子,为什么会有如此大的差别,前者为什么会产生强吸附?谢谢
问题:玄麦甘桔含片中甘草酸的检测:USP拖尾因子是多少呢答案:1.009活动奖励:zengzhengce163(ID:zengzhengce163)sixingxing(ID:v2889187)千层峰(ID:jxyan)http://ng1.17img.cn/bbsfiles/images/2015/12/201512231541_579203_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512231541_579204_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512231542_579205_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512231542_579206_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。玄麦甘桔含片中甘草酸的检测样品制备 制备方法对照品:取甘草酸对照品适量,精密称定,加80%甲醇制成每1 mL含0.2 mg的溶液。分析条件 色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相乙腈:0.1%磷酸溶液=37:63 流速1 mL/min柱温30 ℃检测器UV 250 nm进样量5 μL色谱图对照品http://ng1.17img.cn/bbsfiles/images/2015/12/201512231017_579120_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 21.026 694197 26857 15108.406 1.009 -- *药典要求理论板数按甘草酸峰计算应不低于4000本品种同时使用了Leapsil C18色谱柱,在药典规定条件下进行甘草酸的检测,满足药典要求。

问题:玄麦甘桔胶囊中甘草酸的检测对照品分析中甘草酸的理论塔板数是?答案:15108.406【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币捌道巴拉巴巴巴(注册ID:v3082413)翠湖园(注册ID:hhx050)m3071659(注册ID:m3071659)http://ng1.17img.cn/bbsfiles/images/2016/03/201603171519_587237_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603171519_587238_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================玄麦甘桔胶囊中甘草酸的检测样品制备 制备方法1. 对照品:取甘草酸对照品适量,精密称定,加80%甲醇制成每1 mL含0.2 mg的溶液。2. 供试品:取装量差异项下的本品内容物,研细,混匀,取约1 g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25 mL,称定重量,超声处理(功率250 W,频率33 kHz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相乙腈:0.1%磷酸溶液=37:63 流速1 mL/min柱温30 ℃检测器UV 250 nm进样量5 μL色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/03/201603170956_587210_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 21.026 694197 26857 15108.406 1.009 -- *药典要求理论板数按甘草酸峰计算应不低于4000供试品http://ng1.17img.cn/bbsfiles/images/2016/03/201603170956_587211_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 22.033 991915 35292 14188.640 1.081 -- *药典要求理论板数按甘草酸峰计算应不低于4000本品种同时使用了Leapsil C18色谱柱,在药典规定条件下进行甘草酸的检测,满足药典要求。
草乌薄层喷稀碘化铋钾试液后三种对照品都显什么颜色
问题:玄麦甘桔颗粒中甘草酸的检测:用到了迪马哪几款色谱柱?答案:Platisil ODS,Leapsil C18获奖名单:WUYUWUQIU(ID:wulin321)m3071659(ID:m3071659)sixingxing(ID:v2889187)http://ng1.17img.cn/bbsfiles/images/2016/03/201603231529_587978_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603231529_587979_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。玄麦甘桔颗粒中甘草酸的检测样品制备 制备方法1. 对照品:取甘草酸对照品适量,精密称定,加80%甲醇制成每1 mL含30 μg的溶液,摇匀,即得。2. 供试品:取装量差异项下的本品内容物,研细,混匀,取约1 g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25 mL,称定重量,超声处理(功率250 W,频率33 kHz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相乙腈:0.1%磷酸溶液=37:63 流速1 mL/min柱温30 ℃检测器UV 250 nm进样量5 μL色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/03/201603230951_587901_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 21.299 413351 15834 15232.463 1.016 -- *药典要求理论板数按甘草酸峰计算应不低于4000供试品http://ng1.17img.cn/bbsfiles/images/2016/03/201603230951_587902_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 21.607 706476 26568 14700.090 1.065 -- *药典要求理论板数按甘草酸峰计算应不低于4000本品种同时使用了Leapsil C18色谱柱,在药典规定条件下进行甘草酸的检测,满足药典要求。
配制两个黄芩苷对照品溶液第一个是用50%的甲醇溶解,我感觉这个好难溶,超声了也不见得全部溶解,如果全部溶解配制出来的应该是澄清的吧?该如何处理好呢??第二个要用减压干燥器60度干燥4小时再配制,我觉得涂了凡士林在60度会溶了吧,就不能减压了,我可以直接打开对照品瓶盖直接在烘箱里烘4小时不?这样做会有很大的影响吗?
其实,下面这个应用确切说不应该说是“抢先看”了,因为前几天我们在微信中已经分享了。只是论坛上没有这些应用,于是赶紧发布一下,已经看过的,请飘过哈~~根据国家药典委员会官方网站发布的2015药典“四君子颗粒”公示方法,迪马科技率先进行了此项目的检测,详细应用如下:四君子颗粒中甘草苷、甘草酸铵色谱柱:Diamonsil Plus 5 μm C18, 250 x 4.6 mm流动相:A:乙腈 B:0.05% 磷酸溶液 梯度流速:1.0 mL/min柱温:30℃检测器:UV 237 nm进样量:10 μL对照品溶液http://mmbiz.qpic.cn/mmbiz/sZguaRbQouv2HerhsR5EcrFIEicAd5N1FlfbFzZ7gZibVYc7ZDiafy1aa3adeAXEcoxraNOckqsFwicNxia5F7HG6Gg/640?tp=webp&wxfrom=5药典要求理论塔板数按甘草苷峰计算应不低于5000,而Diamonsil Plus C18 检测的理论塔板数为10814.879,高出药典要求。供试品溶液http://mmbiz.qpic.cn/mmbiz/sZguaRbQouv2HerhsR5EcrFIEicAd5N1F5vNgwbQPicPicjVCqICzDtAZXejMEMxXUMd22ZgMoTXiaK5wFBSIWqdHw/640?tp=webp&wxfrom=5药典要求理论塔板数按甘草苷峰计算应不低于5000,而Diamonsil Plus C18 检测的理论塔板数为10042.045,也高出药典要求。
根据国家药典委员会官方网站发布的2015药典“四君子颗粒”公示方法,迪马科技率先进行了此项目的检测,详细应用如下:四君子颗粒中甘草苷、甘草酸铵色谱柱:Diamonsil Plus 5 μm C18, 250 x 4.6 mm流动相:A:乙腈 B:0.05% 磷酸溶液 梯度流速:1.0 mL/min柱温:30℃检测器:UV 237 nm进样量:10 μL对照品溶液http://mmbiz.qpic.cn/mmbiz/sZguaRbQouv2HerhsR5EcrFIEicAd5N1FlfbFzZ7gZibVYc7ZDiafy1aa3adeAXEcoxraNOckqsFwicNxia5F7HG6Gg/640?tp=webp&wxfrom=5药典要求理论塔板数按甘草苷峰计算应不低于5000,而Diamonsil Plus C18 检测的理论塔板数为10814.879,高出药典要求。供试品溶液http://mmbiz.qpic.cn/mmbiz/sZguaRbQouv2HerhsR5EcrFIEicAd5N1F5vNgwbQPicPicjVCqICzDtAZXejMEMxXUMd22ZgMoTXiaK5wFBSIWqdHw/640?tp=webp&wxfrom=5药典要求理论塔板数按甘草苷峰计算应不低于5000,而Diamonsil Plus C18 检测的理论塔板数为10042.045,也高出药典要求。
目前我们实验室用的维生素A醋酸酯的对照品的供应商断货了,求问一下大家都用的是哪些供应商的对照品?我们也可以去买。我们试用过Sigma的和USP的发现都不行。Sigma的是实际含量和COA上的含量出入较大。USP的是一个混合物有全反式的和CIS的,由于我们不是用的中国药典附录上测定维生素A的方法,所以我们的液相分不开这2种物质,所以也不能用。
益母草与川芎是中药成方制剂中配伍应用频率较高的药对,益母草主要含水苏碱等生物碱类活性成分,川芎主要含阿魏酸等活性成分。对益母草药材及其制剂《中国药典》常以雷氏盐剩余比色法测定益母草总碱含量。在对含益母草及川芎这一药对的制剂进行质量控制时,由于川芎所含生物碱也能与雷氏盐反应生成沉淀,且雷氏盐剩余比色法本身重现性较差,故不宜采用该法。为此,本实验建立了以薄层扫描法测定益母草与川芎合煎液中盐酸水苏碱含量的方法,为制订含有益母草与川芎药对制剂的质量标准提供参考。 1 仪器与试药 CS-9000型双波长薄层扫描仪(日本岛津):939薄层制板器(重庆南岸贝尔德仪器技术厂);定量毛细管(Drummond USA)。盐酸水苏碱对照品(中国药品生物制品检定所,712200105);益母草、川芎(购于安徽毫州)。硅胶G(化学纯,青岛市北区海化干燥剂厂);强酸性阳离子交换树脂(732型,上海化学试剂采购供应站);其余试剂均为分析纯。 2 方法与结果 2.1 益母草与川芎合煎液的制备 取药材100g(益母草-川芎:1:1),加水煎煮2次,每次2h,每次加12倍量水,合并煎液,滤过,滤液浓缩并定容至100mL,即得合煎液样品。 2.2 对照品溶液的制备 取105℃干燥至恒重的盐酸水苏碱对照品适量,精密称定,加乙醇溶解定容,制成1.2mg/mL的对照品溶液。 2.3 供试品溶液的制备 取合煎液样品适量,离心(4O00r/min)5min后,精密量取上清液lmL置小烧杯中,加蒸馏水1OmL,用稀盐酸调pH 1~2,通过已处理好的强酸性阳离子交换树脂柱(内径1cm,长1Ocm)用水洗至流出液无色,弃去水液,再以乙醇-氨水(8:2)150mL洗脱,收集洗脱液,蒸干,残渣加乙醇溶解,定量转移并定容于lmL容量瓶中,摇匀,作为供试品溶液。 2.4 薄层色谱与扫描条件 吸附剂:硅胶G-O.5%CMC-Na薄层板(厚约0.5mm,105℃活化lh,置干燥器中备用):展开剂:丙酮可-无水乙醇-盐酸(10:6:1) :显色剂:喷以改良碘化铋钾-1%FeC13无水乙醇液(5:1)。,冷风吹至斑点显色清晰。 描方式:双波长反射式锯齿扫描:检测波长:λs=525nm,λR=660nm:狭缝:0.4mm×0.4mm: 线形参数SX=3。 2.5 标准曲线的制备 分别精密吸取盐酸水苏碱对照品溶液4.0、6.0、8.0、10.0、12.0μL,分别点于同一薄层板 ,依上述条件展丌,显色,在薄层板上覆盖同样大小的玻璃板,周围用胶布吲定,然后扫描测定各斑点的峰面积积分值。以峰面积积分值(A)对点样量(C)进行回归,得回归方程:A=-37152.7+7579.18C‘r=O.9986。表明在4.8~14.4μg范围内盐酸水苏碱斑点峰面积积分值与点样量呈良好的线性关系。 2.6 稳定性考察 取供试品溶液5μL点样,依上述条件展开,显色,在1h内每隔10min扫描测定1次,结果斑点峰面积积分值RSD为4.6%(n=6),表明斑点在显色后1h内基本稳定。 2.7 精密度考察 精密吸取供试品溶液在同一薄层板上点相同量5点,每点5μL,按上述条件展开,显色,扫描测定,斑点峰面积秋分值RSD为1.33%(n=5),对其中一点连续扫描测定5次,斑点峰面积积分值RSD为0.62%(n=5),表明同板精密度和仪器精密度较好。 2.8 异板精密度试验 取薄层板3块,在每1块薄层板二分别点对照品溶液4、6μL及供试品溶液5μL,依法展开,显色,扫描测定,采用外标两点法计算含量。结果测得供试品溶液中水苏碱含量为1.1308 mg/mL(RSD=3.01%)。 2.9 回收率测定 精密量取已知含量的合煎液样品0.5mL,加入盐酸水苏碱对照品溶液0.5mL,混匀后,按2.3项方法制备供试品溶液。精密吸取供试液5μL,对照品溶液4、6μL,变又点样于同一薄层板上,按上述条件展开、显色、扫描测定,用外标两点法计算。 2.10 样品测定 取合煎液样品,按2.3项 方法制备供试品溶液。精密吸取供试品溶液5μL,对照品溶液4、6μL,分别交叉点于同一薄层板上,按上述条件展开,显色,扫描测定,用外标两点法计算样品中盐酸水苏碱的含量。 3 讨论 在供试品溶液制备中,以甲醇-氨水(8:2) 或乙醇-氨水(8:2) 作洗脱剂,提纯效果均较好。因乙醇比甲醇价廉且安全,建议采用乙醇-氨水(8:2)作为洗脱剂。本实验对洗脱剂用量考察结果表明,用14OmL即可将盐酸水苏碱洗脱完全。为了保证洗脱充分,本实验中确定用150mL洗脱。 盐酸水苏碱为水溶件物质,在薄层板上显色时受湿度影响很大。通过实验考察,优选出其较好的显色方式为:展开后,取出,晾干,丁105℃烘lOmin,再喷显色剂,冷风吹至斑点色清晰。 益母草有效成分水苏碱含量较低且波动大,容易造成含益母草的制剂有效成分低、产品质量不稳定 ,应增加水苏碱含量控制项目。本实验建立的含量测定方法可为制订含益母草及川芎制剂中水苏碱的定量标准提供参考。参考文献:[1]国家药典委员会.中国药典(一部)[S].北京:化学工业出版社,2000.237,455,564.[2]张玲,时延增,于宗渊,等.舣波长薄层扫描法测定益母草LJ服液中水苏碱的含量[J].中国药科大学学报,1996,27(1):16—18.[3]张玲, 宗渊,李国宝,等.7种含益母草中成药中水苏碱的含量测定[J].药物分析杂志,1996,16(3):181—183.
大茴香酸-硫酸荧光体系测定黄芪甲苷刘养清 杜鸣 徐秉玖关键词: 黄芪甲苷; 大茴香酸; 黄芪; 中药复方补阳还五汤; 荧光分光光度法中图分类号: R927.2 R284.1 文献标识码: A 文章编号: 0513-4870(2000)07-0544-03黄芪甲苷(astragaloside)是中药膜荚黄芪Astragalus membranaceus (Fisch.) Bge.和蒙古黄芪Astragalus membranaceus (Fisch.) Bge. var. mongholicus (Bge.) Hsiao的主要活性成分,有抗炎、降压、镇痛、镇静、升高血浆中cAMP水平、促进小鼠再生肝DNA的含量[1,2]以及促进免疫功能等生理活性。黄芪甲苷的测定方法主要有:紫外分光光度法[3,4]、薄层扫描法[5,6]和HPLC法[7,8]。光度法常用香草醛在浓硫酸作用下与甲苷显色反应,空白值较高,干扰严重;薄层扫描法操作繁琐,准确度相对较差。黄芪甲苷仅在200 nm处有末端吸收,对HPLC法不利。黄芪甲苷的荧光分析尚未见报道。本文首次根据在浓硫酸条件下黄芪甲苷与大茴香酸反应产物具有荧光的特性建立了荧光分光光度法测定黄芪甲苷,方法灵敏度高、选择性好、线性范围宽、检出限低、操作简便。可直接用于黄芪生药、中药复方、黄芪制剂以及含药血清等多种样品的测定,无干扰。材料与方法 仪器 日本岛津RF-540型荧光分光光度计。 试剂 黄芪甲苷对照品(中国药品生物制品检定所提供)配成1.0 mg.mL-1的甲醇溶液。2%大茴香酸的无水乙醇溶液,72%硫酸溶液,85%磷酸溶液。所用试剂均为分析纯。 样品及处理 黄芪口服液(上海福达制药有限公司生产,批号980502)。取口服液1.00 mL,加入无水乙醇2.00 mL,离心分离沉淀,上清液蒸干,用甲醇1 mL溶解,备用。补阳还五汤复方汤剂煎煮3次,合并水煎液,分别用石油醚、氯仿、正丁醇萃取4次,每次萃取剂用量为水煎液体积的一半。合并正丁醇相,总体积为800 mL。取正丁醇萃取液2 mL,蒸干,用甲醇2 mL溶解,备用。含药猪血清样品(北京医科大学药学院生药研究室提供):用补阳还五汤复方浸膏连续3 d喂猪,3 d后取血,分离猪血清,取猪血清50 mL,用正丁醇萃取4次,每次萃取剂用量为原血清体积的一半。得含药血清样品100 mL,取正丁醇萃取液4 mL,蒸干,用甲醇2 mL溶解,备用。黄芪生药样品:按文献[3]方法提取分离,甲醇溶样,备用。结果与讨论1 黄芪甲苷反应产物的激发光谱和发射光谱 取1.0 mg.mL-1黄芪甲苷对照品0.1 mL,于5 mL量瓶中,加入2%大茴香酸溶液0.6 mL、72% H2SO4溶液0.8 mL,于60℃水浴中反应20 min,迅速冷却后,用无水乙醇定容至刻度,摇匀,在荧光分光光度计测荧光光谱黄芪甲苷反应产物最大激发波长Ex=320 nm,最大发射波长Em=387 nm。2 大茴香酸用量的影响 取大茴香酸0.1,0.3,0.5,0.6,0.7,0.9 mL按照分析方法操作,选择大茴香酸最佳用量。结果表明大茴香酸用量0.6 mL较合适(图1)。3 稀释液的选择 准确移取1.0 mg.mL-1黄芪甲苷0.1 mL,按照分析方法操作,体积定容时选无水乙醇、甲醇、冰醋酸和水作稀释液,测得其荧光强度(If)分别为77.6,48.4,35.0,1.3。可见无水乙醇对荧光强度影响最小。本文采用无水乙醇作为稀释液。 Fig 1 Effect of amount of anisic acid4 酸的种类和用量的影响 选72% H2SO4, 85% H3PO4及浓HClO4进行实验,结果发现选用72% H2SO4时反应产物的荧光强度最大。对72%硫酸的用量进行选择,结果表明72% H2SO4取0.8 mL为最佳(图2)。 Fig 2 Effect of amount of sulphuric acid5 反应温度的影响 准确移取1.0 mg.mL-1黄芪甲苷对照品0.1 mL,按分析方法内容操作,分别在40,50,60,70,80,90℃和沸水浴中反应20 min,同时做空白。考察荧光强度随温度的变化,结果表明:60℃时反应空白小,荧光强度较高。故实验选择60℃为反应温度较适宜。6 加热时间的影响 将温度控制在60℃,改变加热时间,考察加热时间对反应的影响。结果表明加热时间选20 min为宜。7 反应产物的稳定性 准确移取1.0 mg.mL-1黄芪甲苷对照品0.1 mL,按照分析方法操作,测定荧光强度值,每间隔5 min测定1次,对产物稳定性进行考察。结果表明反应产物在90 min内均稳定。8 工作曲线及检出限 分别准确移取1.0 mg.mL-1黄芪甲苷对照品0.010,0.025,0.050,0.100,0.150和0.200 mL,在最佳实验条件下,测定工作曲线,得回归方程为:Y=-0.4201+1.575X,γ=0.9993。黄芪甲苷浓度在2.0~40 μg.mL-1与荧光强度呈良好的线性关系。检出限为0.02 μg.mL-1。9 干扰考察 为解决基体太浓或基体不一致所造成的影响,在适当稀释溶液后,采用标准加入法测定样品。为考察此反应选择性,利用薄层分离黄芪甲苷[6]后测定样品中其他物质的荧光强度,证明杂质荧光强度与样品总荧光强度的比1.2%。10 样品的测定与回收率实验 取被测样品6份各0.1 mL,依次加入1.0 mg.mL-1黄芪甲苷对照品0.0,0.01,0.02,0.03,0.04和0.05 mL,按照分析方法操作,分别测定了黄芪、复方补阳还五汤、黄芪口服液及猪血清样品中黄芪甲苷的含量,测定结果及回收率实验见表1。SampleContent/%Recovery/%RSD/%HQOL0.210±0.00298.5~101.91.8BYHWT0.280±0.02098.8~102.12.1AMB0.420±0.00498.6~102.02.0PS0.063±0.00198.8~102.41.9黄芪是补阳还五汤的君药,黄芪甲苷定量分析是黄芪中药制剂质量控制的重要指标,本方法灵敏度高,选择性好,操作简便且无干扰,可作为黄芪甲苷的质控方法。基金项目: 九五攀登计划项目杜鸣(北京医科大学药学院分析化学与药物分析研究室,北京 100083 )徐秉玖(北京医科大学药学院分析化学与药物分析研究室,北京 100083 )刘养清(山西师范大学化学系,山西 临汾 041004)收稿日期: 1999-08-03
大家经常做气相需要测对照品溶液,有时对照品溶液的出峰面积在不同时期可能差异较大,大家遇到过这样的问题吗?问题:取甲醇、乙酸乙酯、甲苯适量,精密称定,用DMF溶解稀释定容制成每1ml中约含甲醇60ug、乙酸乙酯100ug、甲苯20ug的混合溶液。那么我们实际称取的对照质量应该在什么范围内是可以接受的?因为称量的差异会导致对照品出峰面积的差异。问题:如何能用天平称准对照试剂的量?(有机溶剂甲醇、乙腈、二氯甲烷等)换算成体积量取还是直接称取?
摘要:建立胡黄连中香草酸和桂皮酸的含量测定方法。方法用双波长扫描法测定胡黄连中香草酸和桂皮酸的含量。结果香草酸。桂皮酸斑点峰面积3Il内稳定,香草酸回收率为103.86%,RSD=1.33%,桂皮酸回收率为103.16%,RSD=1.28%。结论该方法稳定,可行。具有实用性。 关键词:胡黄连 薄层扫描法 香草酸 桂皮酸 胡黄连具有保肝利胆、抗炎、抗真菌等药理作用。胡黄连含胡黄连素、胡黄连苷(I II III)、D-甘露醇、香草酸、肉桂酸、胡黄连醇成分。香草酸和桂皮酸是其中的两种抗菌成分。我们对胡黄连中香草酸、桂皮酸含量建立了薄层扫描法,以达到控制胡黄连的质量,从而为临床疗效提供保证。 1 仪器与试剂 药材:胡黄连,太原市药材公司;仪器:日本岛津CS--9301PC薄层扫描仪;手提式荧光灯(上海固村电光仪器厂);对照品:香草酸对照品(中国药品生物制品检定所);桂皮酸对照品溶液(省药检所提供e=0.604mg/50ml);硅胶GF254(青岛海洋化工厂)所用试剂均为分析纯。 2 实验条件 2.l 薄层层析条件:分别以石油醚-氯仿-丙酮-冰醋酸(10:4.4:10.1);正己烷-乙醚-冰醋酸(5:5:0.1);正己烷-氯仿-乙醚-冰醋酸(5:3:2:0.1)以及氯仿:甲醇(2:1)展开,多次比较发现正己烷。氯仿-乙醚-冰醋酸(5:3:2:0.4)分离效果好。 2.2 测定波长及主要扫描参数,分别对香草酸,桂皮酸对照品斑点在200nm-370nm扫描,在290nm处有最大吸收,350nm处无吸收,固定350nm为参比波长,290nm为测定波长。
我用液相色谱仪测试甘草酸二钾流动相为30乙睛40水可以出峰但是又拖尾,然后我就加了千分之一的三氟乙酸,然后就不出峰了,我想问一下是不是吸附在柱子上了,不加三氟乙酸又可以出峰,我可不可以在加入了三氟乙酸后调高乙睛的比例就可以了。求助各位大神。
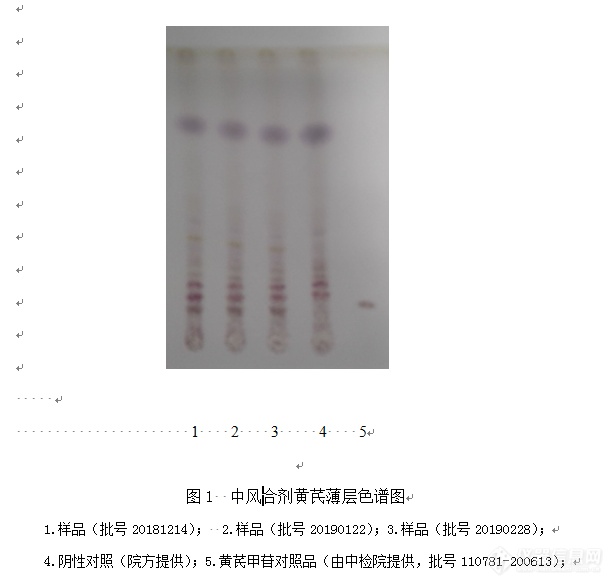
中风合剂(医院制剂质量标准草案起草说明)一.医院制剂用药品的原料(药材)的质量标准草案起草说明:(1)太子参:同正文。 (2)玄参:同正文。 (3)黄芪:同正文。(4)黄精:同正文。 (5)绞股蓝:同正文。 (6)地龙:同正文。(7)僵蚕:同正文。 (8)忍冬藤:同正文。 (9)白芍:同正文。(10)生地黄:同正文。 (11)木瓜:同正文。 二. 临床用药品成品的质量标准草案起草说明:【名称】 中风合剂 Zhongfeng Heji【处方】 太子参150g 玄参150g 黄芪300g 黄精200g 绞股蓝120g 地龙100g 僵蚕100g 忍冬藤150g 白芍120g 生地150g 木瓜120g【制法】以上十一味,用水浸渍30分钟,煎煮两次,第一次1.5小时,第二次1小时,合并煎液,滤过,滤液静置24小时,取上清液浓缩至约800ml,加入甜菊苷、对羟基苯甲酸乙酯和苯甲酸,搅匀,过滤,滤液加水使成1000ml,灌装,灭菌,即得。【性状】 本品为棕褐色液体,味微苦、甜。【鉴别】 处方由11味中药材组成。本标准建立2项薄层色谱鉴别方中2味药材:黄芪、白芍。【鉴别】(1)、(2)均试验了三批样品,并分别与对应的阴性样品进行了比较,均无干扰,且薄层色谱斑点清晰,表明方法可行。(1)系方中黄芪的定性鉴别。方法:取本品20ml,用水饱和的正丁醇振摇提取3次,每次20ml,合并正丁醇液,用1% 氢氧化钠溶液洗涤3次,每次20ml,再用正丁醇饱和的水洗涤至中性,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液,另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(中华人民共和国药典2015年版四部 通则0502)试验,吸取供试品溶液10μl、对照品溶液5μl,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(10:20:11:5)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同顔色的斑点。以黄芪甲苷对照品鉴别方中黄芪,通过阴性对照试验及三批样品的实验观察,阴性无干扰,专属性强,故选黄芪甲苷对照品作为鉴别指标,列入正文,结果见图1。另水饱和的正丁醇提取后,曾用氨试液洗涤去除杂质,但除杂效果不理想,供试品色谱中,在与对照品色谱相应的位置上,斑点干扰大;换用1%氢氧化钠及正丁醇饱和的水溶液洗涤后,获得了理想的薄层色谱条件。[img=,615,587]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111719106767_9782_2166779_3.png!w615x587.jpg[/img](2)[b]系方中白芍的定性鉴别[/b]方法:取本品20ml,用乙醚振摇提取2次,每次20ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取2 次,每次20ml,合并正丁醇液,用水20ml洗涤1次,取正丁醇液,蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。另取芍药苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液。照薄层色谱法(中华人民共和国药典2015年版四部 通则0502)试验,吸取上述两种溶液各lOμl,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(8:1:2:0.1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。以芍药苷对照品鉴别方中白芍,通过阴性对照试验及三批样品的实验观察,阴性无干扰,专属性强,故选芍药苷对照品作为鉴别指标,列入正文,结果见图2。[img=,532,474]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111726009164_2317_2166779_3.png!w532x474.jpg[/img]【含量测定】白芍为方中主药,据《本草拾遗》记载,具有养血柔肝,缓中止痛,敛阴收汗的作用。本文采用HPLC法测定中风合剂中白芍所含有的芍药苷,在测定波长下,阴性无干扰,方法快捷,简便。因此,本文采用HPLC法测定芍药苷的含量,以达到控制中风合剂质量的目的。(一)方法照高效液相色谱法(中华人民共和国药典2015年版四部 0512)测定色谱条件与系统适应性试验 以十八烷基硅烷键合硅胶为填充剂,以乙腈-0.1%磷酸(14:86)为流动相,检测波长为230nm。理论塔板数按芍药苷峰计算应不低于3000。对照品溶液的制备 精密称取芍药苷对照品适量,精密称定,加流动相制成每1ml含芍药苷80μg的对照品溶液,即得。供试品溶液的制备 精密吸取样品1ml,置25 ml量瓶中,用流动相稀释并定容至刻度,摇匀,即为供试品溶液。测定法 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。本品含芍药苷(C23H28O11)的不得少于0.62mg/ml。(二)方法学考察1 仪器与试药戴安U3000高效液相色谱仪;梅特勒XS205DU电子天平;艾科浦超纯水器。中风II号合剂、缺白芍的阴性样品由*********提供。芍药苷对照品(批号110736-201842,含量97.4%)购自中国食品药品生物检定研究院。乙腈为色谱纯;水为超纯水。2 方法与结果2.1 色谱条件色谱柱:Welch Ultimate XB-C18(4.6mm×250mm,5μm);流动相:乙腈-0.1%磷酸(14:86)检测波长:230nm;流速:1.0ml• min-1;柱温:30℃;进样量:10μl理论塔板数:按芍药苷峰计算应不低于3000。2.2 提取方法的选择 在供试品溶液的制备中,进行了直接稀释法、超声法的对比研究,结果两者无显著性差别,从操作简便快捷的角度选择直接稀释法,结果见表2。[img=,657,535]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111734262932_4772_2166779_3.png!w657x535.jpg[/img][img=,690,539]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111734293626_404_2166779_3.png!w690x539.jpg[/img]2.5 空白试验 取对照品溶液、供试品溶液、阴性对照溶液,分别按2.1色谱条件测定,结果见图4。[img=,584,548]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111741164206_471_2166779_3.png!w584x548.jpg[/img][img=,585,401]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111741208226_1968_2166779_3.png!w585x401.jpg[/img][img=,622,365]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111741249466_42_2166779_3.png!w622x365.jpg[/img][img=,631,418]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111741303576_9190_2166779_3.png!w631x418.jpg[/img][img=,665,427]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111741352346_3359_2166779_3.png!w665x427.jpg[/img][img=,615,310]https://ng1.17img.cn/bbsfiles/images/2019/08/201908111741599546_7982_2166779_3.png!w615x310.jpg[/img]本品含芍药苷(C23H28O11)不得少于0.62mg/ml。【功能与主治】 益气养阴、活血通络。用于中风恢复期,半身不遂,肢体麻木,口眼喎斜,舌强语蹇。【用法与用量】 口服,一日2次,一次50ml。或遵守医嘱。【规 格】 100ml/瓶。【贮 藏】 密封。【有效期】2年。
我最近按中国药典2010版方法测定复方甘草口服溶液中甘草酸突然含量测定不上来,原来测定结果能与原料甘草流浸膏中甘草酸基本对应一致,现在会差很大,从柱效、拖尾因子看现在与原先无太大差异,且符合中国药典2010版要求,请教各位同仁,在测定复方甘草口服溶液中甘草酸有什么特别注意点吗?
[size=3]不知大家注意没有,在2010年版药典中,特别是UV-Vis测含量,在“对照品溶液的制备”中,往往是准确指出精密称取的对照品的量,例如,2010年版药典一部第5页,人工牛黄中胆酸的含量测定项下,胆酸对照品溶液的制备:取胆酸对照品12.5mg,精密称定,置25ml量瓶中,加60%冰醋酸溶液使溶解,并稀释至刻度,摇匀,即得(每1ml中含胆酸0.5mg)。而,HPLC或GC等测含量,大多的表述是,如同是第5页,八角茴香中反式茴香脑的含量测定,对照品溶液的制备:取反式茴香脑对照品适量,精密称定,加乙醇制成每1ml含0.4mg的溶液,即得。这两种表述有何不同?[/size]
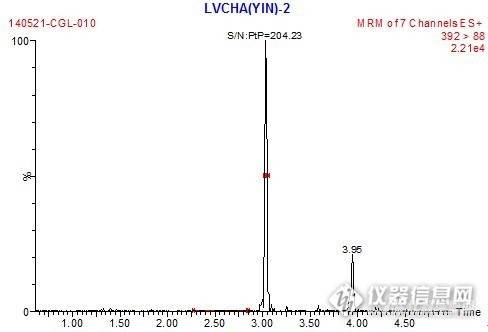
【生活中的仪器分析】样 品:蔬菜、水果、茶叶、茶粉等食品检测项目:草甘膦、草铵膦、氨甲基膦酸参考标准:SN/T 1923-2007检测仪器:a.WATERS液相色谱串联质谱仪:配有电喷雾(ESI)离子源(可用其他品牌作用等效的高效液相色谱质谱仪替代)b.Biotagevacmaster固相萃取仪c.IPRE Qclean PMG草甘膦专用固相萃取柱d.BiotageTurbovap LV 快速浓缩仪e.IKA MS3 涡旋混匀器g.TOMY-MX307离心机g.昆山超声波清洗器实验过程:1.提取及预处理称取2-5g(精确到0.001g)试样于50 mL聚丙烯离心管中,加入100μL内标液,加入20.0 mL水超声提取30min,于10000 r/min离心5min,取1.0 mL上清液于2mL子弹头离心管中,加入100μL酸度调节剂(注A),涡旋混匀,15000r/min离心5 min,待净化。注A:酸度调节剂配制方法:纯水+色谱纯甲醇+盐酸=160+40+13.4(V/V/V)2.固相萃取净化І将PMG-І柱(蓝柱)用2mL甲醇和2 mL 0.5%甲酸淋洗活化并自然滴干,将加入酸度调节剂处理的提取液(2)转移到小柱上,用5mL刻度试管收集流出液(1-2滴/秒),用1.0mL 0.5%甲酸洗柱并真空抽干,合并流出液,用移液枪吸取50%NaOH调pH7-9(用1-14pH试纸,根据样品不同约20-50μL),加水定容到3 mL刻度,混匀,待衍生。3.衍生步骤准确吸取600 μL净化液(3)于2mL子弹头离心管中,加入200 μL 5%硼砂溶液,边涡旋,边加入200 μL 25g/L FMOC-Cl乙腈溶液(注B),放置10min,加入50 μL甲酸,涡旋混匀,15000 r/min离心5min,吸取上清液准备过PMG-ІІ柱。注B:25 g/L FMOC-Cl乙腈溶液配制方法:称取0.25 gFMOC-Cl,溶解于10 mL色谱纯乙腈中。4.固相萃取净化ІІ 将PMG-ІІ柱(红柱)用2 mL甲醇和2 mL 0.5%甲酸淋洗并自然滴干,将上清液过PMG-ІІ柱,用3 mL水淋洗小柱,真空抽干5-10 min,再加入2 mL正己烷淋洗小柱,滴干后真空抽干5 min,最后用5 mL 5%氨水/甲醇洗脱小柱(1-2 mL/min)并用5 mL刻度试管收集流出液,45℃,氮气吹至近干,用20%乙腈定容1.0 mL,涡旋混匀,过0.2 μm PTFE膜后上机测试。5.测定5.1色谱条件a.色谱柱:Waters BEH-C18,1.7 μm,2.1 mm×100 mm;b.流动相:5mmol/L乙酸铵:乙腈梯度洗脱,梯度表见表1; 表1 流动相及梯度 时间(min)流速(mL/min)5mmol/L乙酸铵(%)乙腈(%)00.3901020.362384.40.362384.50.35956.50.35956.60.390109.00.39010c.检测器:串联四极杆质谱仪;d.柱温:35℃;e.进样量:10 μL。5.2质谱分析条件a)电离源:电喷雾正离子模式;b)毛细管电压:3.50KV;c)源温度:120℃;d)脱溶剂气温度:400℃;e)脱溶剂气流量:700L/h;f)碰撞室压力:2.7í10-3mbar;g)特征离子及参数见表2。 表 2 草甘膦和氨甲基膦酸的主要特征离子 化合物保留时间(min)母离子+(m/z)锥孔电压(V)子离子(m/z)碰撞能量(eV)草甘膦1.32392.215*88.02515214.01
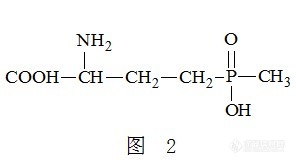
关于”新版GB 2763 食品安全国家标准 规定茶叶中限量农残草甘膦和草铵膦项目“的检测研究 一、研究意义及现状 随着新版GB 2763 食品安全国家标准的不断更新及发布实施,草甘膦和草铵膦已被明确列为茶叶中农药残留强检(必检)项目,草甘膦在茶叶中的限量为1mg/kg,草铵膦在茶叶中的限量为0.5mg/kg。同时,草甘膦和草铵膦也成为中国茶叶出口国外的检测项目(来源于中华人民共和国商务部),且已成为越来越严的限量指标。 文献(2013年农药行业预测和草甘膦市场机遇分析,杨益军,农药市场信息,2013.03)报道,除草剂草甘膦因其高效、广谱、低毒等特性使其被广泛应用,未来需求量也将大幅增加。但草甘膦的使用容易使植物产生抗性(IARC国际研究机构发布报告称草甘膦很可能对人类致癌),而草铵膦可克服该缺陷,现已有学者(草铵膦、百草枯、草甘膦对非耕地杂草的防效比较,凌进,农药,2014年第53卷第8期,613-615)对草铵膦和草甘膦的除草性能进行了研究,确证了草铵膦代替草甘膦的可行性。 因草甘膦和草铵膦为广谱除草剂,被广泛应用于农业、林业及园艺的栽培。我国作为农业大国,其茶叶产量世界第一、出口量世界第二,草甘膦和草铵膦的生产和使用量都位居世界前列(草甘膦 草铵膦及其代谢产物的检测方法,李小娟、周信康、孟品佳,公共安全中的化学问题研究进展)。同时,我单位对西南茶叶原料主产区进行了初步调研,进一步确认茶农使用草甘膦和草铵膦农药的现状。 随着草甘膦和草铵膦除草剂使用量的日益增大,使其常被发现存在于环境水样、土壤及植物中,这样长期积累会引起环境污染,从而对人类健康造成严重威胁。草甘膦和草铵膦结构类似,且均含有膦酸基、羟基、氨基,是极强的两性化合物,易溶于水,难挥发。鉴于草甘膦和草铵膦特殊的物化性质和茶叶基质自身的复杂性,无论国内外,茶叶中草甘膦和草铵膦同时检测的标准还未见发布。 目前,可用于检测草甘膦和草铵膦农药残留量的主要方法有液相色谱法,柱前衍生后气相色谱法、气相色谱-质谱法及液相色谱-质谱/质谱法。 快速发展起来的超高效液相色谱-质谱联用技术,具有检测灵敏度高、适用范围广、分析速度快和能有效排除复杂基质产生的干扰等优点,当今已成为检测型实验室检测农残的首选。然而,若采用液质质直接测定草甘膦和草铵膦,则仪器响应较低,无法满足茶叶中草甘膦和草铵膦农药残留量检测的要求。 近两年来,已有研究文献陆续发表,用柱前衍生-液相色谱串联质谱法检测。笔者结合其文献研究结果,对茶叶中草甘膦和草铵膦农药残留量的检测方法系统地进行研究,采用9-芴甲氧羰酰氯(FMOC-Cl)作为常用衍生剂,在硼酸盐缓冲盐溶液的条件下,能与草甘膦和草铵膦的提取液发生衍生反应,形成衍生产物,衍生产物注入UPLC进行色谱洗脱分离,采用串联质谱探测响应信号,外标法直接快速定量茶叶中的草甘膦和草铵膦的含量。二、液质质检测分析原理 质谱原理是先将物质离子化,按离子的质荷比分离,然后测量各种离子谱峰的强度而实现分析目的的一种分析方法。液质联用是将色谱的分离能力与质谱强大的定性功能结合起来,实现对复杂混合物更准确的定量和定性分析,简化样品的前处理流程,使样品分析更简便。主要针对不挥发性、极性、热不稳定、大分子量等化合物的分析测定。液质联用检测技术灵敏度高,且串联质谱(三重四级杆)定性准确,可有效杜绝微量甚至痕量物质分析时的假阳性现象,常用于目标物质的痕量分析。 采用柱前衍生-液相色谱串联质谱法检测茶叶中的草甘膦和草铵膦有以下优势: 1)、灵敏度高、线性好、检出限低(可达ng/mL级及其以下); 2)、定量结果准确、稳定、重复性好; 3)、实验操作简单、步骤少、耗时短、分析速度快、检测效率高; 4)、实验试剂无污染、无毒、安全; 5)、有效减弱基质对目标物检测的影响。三、茶叶中草甘膦和草铵膦农药残留量检测的前处理试验 茶叶经GB/T8303磨碎、过筛制得待测茶样(发酵茶应先低温去除水分,使样品易于磨碎); 准确称取已磨碎处理过的茶样1g(精确至0.001g)置于80mL具盖离心管中,加入10mL水,涡旋混匀静置,加入2mL二氯甲烷,混匀,超声提取或回旋振荡10min,低速离心机4500r/min离心5min,取上清液,制得提取上清液; 注意:若茶样为新采摘的鲜叶,则称取约5g鲜叶于研钵中,加入30mL水,研磨约10min,将其转入离心管中,用10mL水洗涤研钵后转移至离心管,重复洗涤一次,再次加入10mL二氯甲烷于离心管,均质至混匀,4500r/min离心10min,取上清液,制得提取上清液; 将提取上清液用净化柱CAX、C18,以及活性炭小柱等进行比对试验,确定以C18小柱净化提取液,制得提取净化液; 通过缓冲液浓度、衍生液浓度、衍生液用量、缓冲液用量、净化液用量,衍生时间等条件试验,得出最优衍生试验参数为缓冲液浓度为50g/L,衍生液浓度20g/L,衍生液用量:缓冲液用量:净化液用量的体积比为1:1:1,衍生时间为约3h,衍生液过0.22μm的有机滤膜后进样。四、茶叶中草甘膦和草铵膦农药残留量检测的衍生机理 茶叶中草甘膦和草铵膦农药残留量的衍生机理为:在硼酸钠缓冲盐溶液条件下,草甘膦(分子结构如图1所示)和草铵膦(分子结构如图2所示)中R-NH-R’的-H被FMOC-Cl(分子结构如图3所示)中的FMOC-取代,生成 http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414494663_01_0_3.png,得到衍生目标产物草甘膦衍生物和草铵膦衍生物。 其中,草甘膦分子结构图,见图1;草铵膦分子结构图,见图2;9-芴甲氧羰酰氯(FMOC-Cl)分子结构图,见图3;草甘膦和草铵膦与9-芴甲氧羰酰氯(FMOC-Cl)的衍生机理图,见图4。http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414424276_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414430242_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414432007_01_2275853_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507061123_553629_2275853_3.png五、茶叶中草甘膦、草铵膦农残衍生物在质谱中的裂解机理1、茶叶中草甘膦农残衍生物在质谱中的裂解机理 通过对草甘膦衍生物在串联质谱中的裂解机理进行系统的分析研究,可探索出草甘膦衍生物的裂解机理为:首先草甘膦衍生物裂解为离
有没有做过甘草浸膏的,测甘草苷的对照品,为什么甘草苷后面出杂峰哪[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221438425145_6044_3461163_3.jpeg[/img]

[align=center][b]中风Ⅰ号合剂[/b][/align][align=center][b]医院制剂用药品的原料(药材)和成品的质量标准草案[/b][/align][b]一. 医院制剂用药品的原料(药材)的质量标准草案:[/b](1)大黄:本品为蓼科植物掌叶大黄Rheumpalmatuml.、唐古特大黄Rheumtanguticum.Maxim.exBalf. 或药用大黄RheumoffcihaleBaill. 的干燥根和根茎。秋末茎叶枯萎或次春发芽前采挖,除去细根,刮去外皮,切瓣或段,绳穿成串干燥或直接干燥。应符合中华人民共和国药典2015年版一部23页大黄项下的有关规定。(2)钩藤:为茜草科植物钩藤 [i]Unacaria rhynchophylla[/i](Miq.)Miq.ex Havil.、大叶钩藤[i] Uncaria macrophylla[/i] Wall.、毛钩藤[i]Uncaria hirsuta[/i] Havil.、华钩藤 [i]Uncaria sinensis[/i](Oliv.)Havil.或无柄果钩藤[i]Uncaria sessilifructus[/i] Roxb.的干燥带钩茎枝。秋、冬二季采收,去叶,切段,晒干。主产于[color=#333333]浙江、福建、广东、广西[/color][color=#333333]等省。[/color]应符合中华人民共和国药典2015年版一部257页钩藤项下的有关规定。(3)白芍:为毛莨科植物芍药Paeonia lactiflora PalL.的干燥根。夏、秋二季采挖,洗净,除去头尾和细根,置沸水中煮后除去外皮或去皮后再煮,晒干。主产于[color=#333333]浙江、安徽、四川等省。[/color]应符合中华人民共和国药典2015年版一部105页白芍项下的有关规定。 (4)夏枯草:为唇形科植物夏枯草Prunella vulgarisL.的干燥果穗。夏季果穗呈棕红色时采收,除去杂质,晒干。主产于[color=#333333]江苏、安徽、浙江、河南[/color]等省。应符合中华人民共和国药典2015年版一部280页夏枯草项下的有关规定。(5)浙贝母:为百合科植物浙贝母Fritillariathunbergii Miq.的干燥鱗茎。初夏植株枯萎时采挖,洗净。大小分开,大者除去芯芽,习称“大贝”;小者不去芯芽,习称“珠贝”。分别撞擦,除去外皮,拌以锻过的贝壳粉,吸去擦出的浆汁,干燥;或取鱗茎,大小分开,洗净,除去芯芽,趁鲜切成厚片,洗净,干燥,习称“浙贝片”。主产于[color=#333333]浙江、江苏、湖南[/color]等省。应符合中华人民共和国药典2015年版一部292页浙贝母项下的有关规定。(6)地龙:为钜蚓科动物参环毛蚓Pheretima aspergillum(E.Perrier)、通俗环毛蚓Pheretima vu1garis Chen、威廉环毛蚓Pheretima guillelmi(Michaelsen)或栉盲环毛蚓Pheretima pectinifera Michaelsen的干燥体。前一种习称“广地龙”,后三种习称“沪地龙”。广地龙春季至秋季捕捉,沪地龙夏季捕捉,及时剖开腹部,除去内脏和泥沙,洗净,晒干或低温干燥。[color=#333333]广地龙[/color][i]主产于[/i]广东、海南、广西、福建等省。沪[i]地龙主产于[/i]上海、浙江等省。应符合中华人民共和国药典2015年版一部122页地龙项下的有关规定。(7)石决明:为鲍科动物杂色鲍Haliotis diversicolor Reeve、皱纹盘鲍Haliotis discus hannai Ino、羊鲍Haliotis ovinaGmelin、澳洲鲍Haliotis ruber(Leach)、耳鲍Haliotis asinina Linnaeus或白鲍Haliotislaevigata(Donovan)的贝壳。夏、秋二季捕捞,去肉,洗净,干燥。主产于[color=#333333]浙江、福建、台湾、广东、海南、广西[/color]等省。应符合中华人民共和国药典2015年版一部91页石决明项下的有关规定。(8)鲜竹沥:为禾木科植物粉绿竹Phyllostachys glaucaMcClure、净竹Phyllostachysnuda McClure及同属数种植物的鲜杆经加热后自然沥出的液体,煮沸后,加适量防腐剂制得。主产于四川、江西等省。应符合中华人民共和国卫生部药品标准中药材第一册99页鲜竹沥项下的有关规定。[b]二.医院制剂用药品的成品的质量标准草案:[/b][align=center][b]中风Ⅰ号合剂[/b][/align][align=center][/align][b]【处方】 [/b]大黄60g 钩藤120g 白芍100g 夏枯草150g浙贝母90g 地龙100g 石决明240g 鲜竹沥100ml[b]【制法】 [/b]以上八味药材,除鲜竹沥,其余七味用水浸渍30分钟,煎煮两次,第一次1.5小时,第二次1小时,合并煎液,滤过,滤液静置24小时,取上清液浓缩至约800ml,加入鲜竹沥、甜菊苷、对羟基苯甲酸乙酯和苯甲酸,搅匀,过滤,滤液加水使成1000ml,灌装,灭菌,即得。[b]【性状】 [/b]本品为棕褐色液体,味微苦、甜。[b]【鉴别】 [/b](1)取本品20ml,加盐酸2ml,水浴加热30分钟,放冷,用乙醚振摇提取3次,每次25ml,合并乙醚液,挥干,残渣加甲醇1ml使溶解,作为供试品溶液。取大黄素对照品、大黄酚对照品及大黄酸对照品,加甲醇分别制成每1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(中华人民共和国药典2015年版四部 通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以石油醚(30〜 60°C)-甲酸乙酯-甲酸(15:5:1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。(2)取本品20ml,用乙醚振摇提取2次,每次20ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取2 次,每次20ml,合并正丁醇液,用水20ml洗涤1次,取正丁醇液,蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。另取芍药苷对照品,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。照薄层色谱法(中华人民共和国药典2015年版四部 通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(8:1:2:0.1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[b]【检查】 相对密度[/b] 应不低于1.02(中华人民共和国药典2015年版四部通则0601)。[b] pH值[/b] 应为4.0~6.0(中华人民共和国药典2015年版四部 通则0631)[b] 其他 [/b]应符合合剂项下有关的各项规定(中华人民共和国药典2015年版四部 通则0181)。[b]【含量测定】 [/b]照高效液相色谱法(中华人民共和国药典2015年版四部通则0512)测定[b]色谱条件与系统适应性试验[/b] 以十八烷基硅烷键合硅胶为填充剂,以乙腈-0.1%磷酸(14:86)为流动相,检测波长为230nm。理论塔板数按芍药苷峰计算应不低于3000。[b]对照品溶液的制备 [/b]精密称取芍药苷对照品适量,精密称定,加流动相制成每1ml含芍药苷80μg的对照品溶液,即得。[b]供试品溶液的制备[/b] 精密吸取样品1ml,置25ml量瓶中,用流动相稀释并定容至刻度,即为供试品溶液。[b]测定法[/b] 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。本品含芍药苷(C[sub]23[/sub]H[sub]28[/sub]O[sub]11[/sub])不得少于0.35mg/ml。[b]【功能与主治】 [/b]平肝熄风、化痰通腑。用于各类急性期中风,半身不遂,肢体麻木,口眼喎斜,舌强语蹇。[b]【用法与用量】 [/b]口服,一日2次,一次50ml。或遵守医嘱。[b]【规 格】 [/b] 100ml/瓶。[b]【贮 藏】 [/b] 密封。[b]【有效期】 [/b]2年。[align=center][b]中风Ⅰ号合剂[/b][/align][align=center][b]医院制剂用药品的原料(药材)和成品的[/b][/align][align=center][b]质量标准草案起草说明[/b][/align][b]一.医院制剂用药品的原料(药材)的质量标准草案起草说明:[/b](1)大黄:同正文。 (2)钩藤:同正文。 (3)白芍:同正文。(4)夏枯草:同正文。 (5)浙贝母:同正文。 (6)地龙:同正文。(7)石决明:同正文。 (8)鲜竹沥:同正文。[b]二.临床用药品成品的质量标准草案起草说明:【名称】 [/b]中风Ⅰ号合剂 ZhongfengYihao Heji[b]【处方】[/b] 同正文。[b]【制法】 [/b]同正文。[b]【性状】 [/b]同正文。[b]【鉴别】 [/b]处方由8味中药材组成。本标准建立2项薄层色谱鉴别方中2味药材:大黄、白芍。【鉴别】(1)、(2)均试验了三批样品,并分别与对应的阴性样品进行了比较,均无干扰,且薄层色谱斑点清晰,表明方法可行。[b](1)系方中大黄的定性鉴别。[/b]以大黄素、大黄酚、大黄酸对照品鉴别方中大黄,通过阴性对照试验及三批样品的实验观察,阴性无干扰,专属性强,故选大黄素、大黄酚及大黄酸对照品作为鉴别指标,列入正文(见图1)。[img=,596,504]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010912419216_3978_2166779_3.png!w596x504.jpg[/img][b](2)系方中白芍的定性鉴别。[/b]以芍药苷对照品鉴别方中白芍,通过阴性对照试验及三批样品的实验观察,阴性无干扰,专属性强,故选芍药苷对照品作为鉴别指标,列入正文(见图2)。[img=,690,649]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010913524007_844_2166779_3.png!w690x649.jpg[/img][b]【含量测定】[/b]白芍为方中主药,据《本草拾遗》记载,具有[color=#333333]养血柔肝,缓中止痛,敛阴收汗[/color]的作用。本文采用HPLC法测定中风I号合剂中白芍所含有的芍药苷,在测定波长下,阴性无干扰,方法快捷,简便。因此,本文采用HPLC法测定芍药苷的含量,以达到控制中风I号合剂质量的目的。[b](一)方法[/b]照高效液相色谱法(中华人民共和国药典2015年版四部 0512)测定[b]色谱条件与系统适应性试验[/b] 以十八烷基硅烷键合硅胶为填充剂,以乙腈-0.1%磷酸(14:86)为流动相,检测波长为230nm。理论塔板数按芍药苷峰计算应不低于3000。[b]对照品溶液的制备[/b] 精密称取芍药苷对照品适量,精密称定,加流动相制成每1ml含芍药苷80μg的对照品溶液,即得。[b]供试品溶液的制备[/b] 精密吸取样品1ml,置25 ml量瓶中,用流动相稀释并定容至刻度,摇匀,即为供试品溶液。[b]测定法[/b] 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。本品含芍药苷([color=#333333]C[sub]23[/sub]H[sub]28[/sub]O[sub]11[/sub][/color])的量不得少于0.35mg/ml。[b](二)方法学考察1 仪器与试药[/b]戴安U3000高效液相色谱仪;梅特勒XS205DU电子天平;艾科浦超纯水器。中风I号合剂由福建省南平市人民医院制剂室提供。芍药苷对照品(批号110736-201842,含量97.4%)购自中国食品药品生物检定研究院。乙腈为色谱纯;水为超纯水。[b]2 方法与结果2.1 色谱条件[/b]色谱柱:Welch Ultimate XB-C18(4.6mm×250mm,5μm);流动相:乙腈-0.1%磷酸(14:86)检测波长:230nm;流速:1.0mlmin[sup]-1[/sup];柱温:30 ℃;进样量:10μl理论塔板数:按芍药苷峰计算应不低于3000。[b]2.2 提取方法的选择 [/b]在供试品溶液的制备中,进行了直接稀释法、超声法的对比研究,结果两者无显著性差别,从操作简便快捷的角度选择直接稀释法,结果见表2。 表2 芍药苷不同提取方法含量测定结果比较 [table=594][tr][td] [align=center]提取方法[/align] [/td][td] [align=center]芍药苷含量(mg/ml)[/align] [/td][td] [align=center]平均含量(mg/ml)[/align] [/td][/tr][tr][td=1,2] [align=center]稀释法[/align] [/td][td] [align=center]0.6890[/align] [/td][td=1,2] [align=center]0.69[/align] [/td][/tr][tr][td] [align=center]0.6888[/align] [/td][/tr][tr][td=1,2] [align=center]超声法[/align] [/td][td] [align=center]0.6892[/align] [/td][td=1,2] [align=center]0.69[/align] [/td][/tr][tr][td] [align=center]0.6890[/align] [/td][/tr][/table][b]2.3 溶液的制备[/b]2.3.1对照品储备液的制备 精密称取芍药苷对照品10mg,置10ml量瓶中,用甲醇溶解并稀释至刻度,制得对照品储备液(0.974g/L芍药苷)。2.3.2 供试品溶液的制备 精密吸取样品1ml,置25ml量瓶中,用流动相稀释并定容至刻度,摇匀,即为供试品溶液。2.3.3 阴性对照溶液的制备 按处方比例制备不含芍药苷的阴性样品,同2.3.2制备方法制备阴性对照溶液。[b]2.4 线性关系考察[/b]将芍药苷对照品储备液逐步稀释,得到浓度分别为4.87,9.74,24.35,48.70,73.05,97.40μg/ml六个浓度的系列标准溶液,进样测定,结果见表3 [table][tr][td=3,1] [align=center]表3 芍药苷线性关系测定结果[/align] [/td][/tr][tr][td] [align=center]进样体积(μl)[/align] [/td][td] [align=center]芍药苷浓度(μg/ml)[/align] [/td][td] [align=center]峰面积(mAU*min)[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]4.87[/align] [/td][td] [align=center]1.105[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]9.74[/align] [/td][td] [align=center]2.252[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]24.35[/align] [/td][td] [align=center]5.853[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]48.70[/align] [/td][td] [align=center]11.909[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]73.05[/align] [/td][td] [align=center]17.511[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]97.40[/align] [/td][td] [align=center]23.240[/align] [/td][/tr][/table]以峰面积(Y)为纵坐标,以芍药苷浓度(X)为横坐标绘制标准曲线。结果表明,芍药苷在4.87~97.40μg/ml的范围内线性关系良好(见图3)[img=,611,350]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916394318_5586_2166779_3.png!w611x350.jpg[/img][img=,650,539]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916445229_4881_2166779_3.png!w650x539.jpg[/img][img=,631,383]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916515496_9756_2166779_3.png!w631x383.jpg[/img][img=,618,717]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916572812_7861_2166779_3.png!w618x717.jpg[/img][img=,646,703]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010917032958_603_2166779_3.png!w646x703.jpg[/img][b]【功能与主治】 [/b] 同正文。[b]【用法与用量】 [/b] 同正文。[b]【规 格】 [/b] 同正文。[b] 【贮 藏】 [/b]同正文。[b]【有效期】 [/b]同正文。
求2010版《药典》甘草浸膏含甘草酸、甘草苷两个成分的图谱http://simg.instrument.com.cn/bbs/images/brow/em09512.gif做了一下,没做出来。感觉不太对。有做过的给点经验。谢谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP