氯唑沙宗对照品和对乙酰氨基酚对照品的出峰时间分别是多久?
氯化乙酰胆碱 对照液的配制前需干燥至恒重50℃干燥了3小时,放入干燥器冷却后,发现称量杯内壁出现小液珠刚取出来的时候并未见液珠。请问各位大虾,该如何干燥?我不确定液珠是否是水珠。
乙酰二茂铁的合成目的原理实验目的 1 通过乙酰二茂铁的制备,了解用Friendel-Crafts酰基化反应制备非苯芳酮的原理和方法。2 学习柱色谱分离提纯产品和薄层色谱跟踪反应进程的原理和操作方法。实验原理 二茂铁又名双环戊二烯基铁,是由2个环戊二烯负离子和一个二价铁离子键合而成。一般认为,以乙酸酐为酰化剂,三氟化硼,氢氟酸,磷酸为催化剂,主要生成一元取代物;如用无水三氯化铝为催化剂,酰氯或酸酐为酰化剂,当酰化剂与二茂铁的摩尔比为2∶1时,反应产物以1,1′-二元取代物为主。二茂铁及其衍生物的分离最好是用层析法。本实验用柱色谱分离提纯产品,可用薄层色谱法跟踪反应进程,柱色谱和薄层色谱均属于吸附色谱,柱色谱分离提纯是根据二茂铁,乙酰二茂铁和1,1′-二乙酰基二茂铁对活性氧化铝吸附能力的差异而进行分离提纯。用薄层色谱跟踪反应进程,根据二茂铁和乙酰二茂铁的斑点大小可以了解乙酰化反应的进程。仪器药品 5ml圆底烧瓶,克莱森接头,干燥管,电磁加热搅拌器,30cm色谱柱(自制),30×100mm载玻片,离心试管50ml烧杯,玻璃钉漏斗,吸滤瓶,锥形瓶,氮气袋,250ml烧杯二茂铁,乙酸酐,85%H3PO4,25%NaOH,二氯甲烷,棉花,洗净的砂,Ⅲ级活性氧化铝,己烷,醇,硅胶,0.5%羚甲基纤维素,干燥氮气。过程步骤 一、乙酰二茂铁的制备称取100mg(0.54mmol)二茂铁,放入5ml圆底烧瓶中,加入2.0ml醋酸酐。装上带有干燥管的克莱森接头。水浴温热并搅拌使二茂铁溶解。移去水浴,打开塞子迅速加入3ml 85% H3PO4,使反应液变成深红色,室温下搅拌1.5h,在反应期间定期用毛细管在液面上吸取2滴左右反应液放入具塞小试管中,假如10滴二氯甲烷,所得溶液用薄层色谱法展开,以了解反应进程。当二茂铁的斑点很浅时,表示反应基本完成。将反应液滴入盛有1g碎冰5ml烧杯中,滴加25%NaOH中和恰至碱性,得到大量桔黄色沉淀。充分冷却后抽滤,1ml冷水分几次洗涤沉淀,抽干,干燥后称重约110~120mg。二、乙酰基二茂铁的柱色谱法分离(1)干法装柱将粗产品溶于0.5ml二氯甲烷加入300mgⅢ级活性氧化铝,振荡均匀得浆状物。在通风橱中,在干燥氮气下除去溶剂至恒重,得到松散的颗粒状物,精确称取1/2用作柱色谱分离。将自制的1.5×30cm色谱柱洗净,干燥,柱底铺一层玻璃棉或脱脂棉,再铺一层约5~8mm厚的砂,填平。称取5gⅢ级活性的中性氧化铝(60~80目),通过漏斗将氧化铝装入柱管内,轻敲柱管,使之填均匀。将精确称得含有1/2产品重的氧化铝装入柱内,顶部盖一层约5mm厚的砂子,使氧化铝顶端和砂子上层保持水平。(2)洗脱用己烷作洗脱剂从柱顶加入,缓慢滴入己烷逐渐展开得到黄色、橙色分离的色谱带。黄色的二茂铁带首先从柱下流出,用己称重的锥形瓶收集洗脱溶液。当黄色谱带完全洗脱下来时,改用体积比为1∶1的二氯甲烷己烷混合物洗脱,同时橙色带往下移动,逐渐改变溶剂的比例到体积比9∶1二氯甲烷己烷混合溶剂时,则将橙色色谱带完全洗脱下来,用另一只已称重的锥形瓶收集洗脱液。最后改用体积比为9∶1二氯甲烷甲醇洗脱时,可以看到很淡的,很少量的,棕色色带向下移动,将该洗脱液另行收集。(3)收集产品在通风橱内,各组分洗脱液分别在水浴上蒸馏,回收溶剂。浓缩后的溶液放置冷却析出结晶,将产品放在盛有石蜡片的干燥器内至恒重。可回收到未反应的二茂铁20~22mg;得到乙酰二茂铁80~90mg 1,1′-二乙酰基二茂铁少于2mg。分别测定熔点。注意事项1.二茂铁需经升华或用石油醚(30~60℃)重结晶纯化。2.仪器应是充分干燥的。3.乙酸酐是临用前经重新蒸馏的。4.吸附剂的活性与其含水量的关,含水量越低,活性越高。氧化铝放入高温炉中(300~400℃)烘3h得无水物即Ⅰ级氧化铝。Ⅲ级氧化铝可用Ⅰ级活性氧化铝加入重量的6%的水而得到。如所用氧化铝活性过强会使产品不易洗脱,浪费较多的溶剂。5.这里是考虑到柱色谱的容器。一般粗产品重75mg以上都仅取1/2作柱色谱分离。6.二茂铁易升华,故测熔点时要封管。熔点的文献值:二茂铁为173℃,乙酰二茂铁为85℃,1,1ˊ-乙酰基二茂铁为130℃。分析思考1. 二茂铁乙酰化反应的机理怎样?2. 怎样利用薄层层析判断乙酰化反应的进程?3. 乙酰二茂铁在石油醚和乙醚中溶解度哪个更大?为什么?4. 柱层析分离二茂铁衍生物时,如何选择展开的溶剂? [img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52002_1632583_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52003_1632583_3.gif[/img]
请各位大侠帮忙看看,下面这份标准是否存在错误。样品名称:乙酰半胱氨酸(原料)检验项目:有关物质方法:高效液相色谱法色谱条件与系统适应性试验:采用磺酸型强阳离子交换柱,以水(用0.05mol/L的硫酸溶液调节pH值为3.0)为流动相,检测波长:210nm。理论板数按乙酰半胱氨酸峰计算应不低于1200。测定法:取本品,加流动相制成每1ml中约含0.3mg的溶液,作为供试品溶液;精密量取供试品溶液1.0ml,置100ml容量瓶中,加水稀释至刻度,摇匀,作为对照溶液;精密量取对照溶液20ul注入氨基酸分析仪,调节检测灵敏度,使主成分峰高为满量程的10%-25%;再取供试品溶液20ul注入氨基酸分析仪,记录色谱图至主成分峰保留时间的2倍,按面积归一化法计算,杂质总量不得过1.0%。以上样品可以直接用高效液相色谱法紫外检测器进行检验,而不经过氨基酸分析仪吗?
样品为复方维生素,其中一项是核黄素磷酸钠,没找到核黄素磷酸钠的对照品,故用的核黄素对照品样品制备:先用水溶,然后用流动相稀释,流动相弱酸性做出的结果比标示量低了很多啊用核黄素对照品代替核黄素磷酸钠对照品,请问结果可信吗?
今天做了一个感冒灵颗粒的鉴别,样品点能与对照品扑尔敏点对上,但是就是与对照品对己酰氨基酚难对上,药典要求是15ul,我把对照品的量减少了,样品的量加大了一倍,也不是很容易找到,请问这是什么原因?
在抗生素类的标准物质使用时,经常会遇到标准品和对照品的概念。关于这二者的区别,现在比较流行的说法是在做HPLC时使用的标准物质应为对照品。摘录典型观点如下:[B]“标准品都是按效价单位(或μg)计,以国际标准品进行标定。标准品的标示量是按生物活性来计算的,不是按纯度来标示,此种标示法对单组分或多组分物质均适用,尤适用于多组分物质,如乙酰螺旋霉素标准品,是由4种有效成分组成,若欲于一个纯度来标示其含量是不可能的,但用效价(即生物活性)来标示是可行的;对照品的标示量则必定是某单一组分的纯度指标。所以日常工作中,标准品和对照品在定量时是不可相互替代的。以罗红霉素为例,现今是国家标准品与对照品并存,以抗生素微生物检定法测其含量时,必须使用罗红霉素标准品;但以HPLC法测定其含量时,又必须使用罗红霉素对照品,不可混淆。”[/B]但是我见过一些行业标准,比方说HPLC测土霉素残留中,在说到标准液的配制时,写得就是“土霉素标准品”。难道这里面的“标准品”是“对照品”的错误用法?[em0716] 请大家发表一下看法
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。
项目概况宜春市检验检测中心试剂及耗材和标准品及对照品采购项目(第二次)(第二包:标准物质、对照品) 招标项目的潜在投标人应在 江西省公共资源交易网 获取招标文件,并于 2023年02月09日 09点00分 (北京时间)前递交投标文件。一、项目基本情况:项目编号:明月-YC2022-042-2-1项目名称:宜春市检验检测中心试剂及耗材和标准品及对照品采购项目(第二次)(第二包:标准物质、对照品)采购方式:公开招标预算金额:400000.00 元最高限价:400000.00采购需求:[table=100%][tr][td]采购条目编号[/td][td]采购条目名称[/td][td]数量[/td][td]单位[/td][td]采购预算(人民币)[/td][td]技术需求或服务要求[/td][/tr][tr][td][font=inherit]宜购2022F000814837[/font][/td][td][font=inherit]2022年食品药品监管中央及省级补助资金(标准品、对照品)[/font][/td][td][font=inherit]1[/font][/td][td][font=inherit]批[/font][/td][td][font=inherit]400000.00元[/font][/td][td][font=inherit]详见公告附件[/font][/td][/tr][/table]合同履行期限:详见招标文件本项目不接受联合体投标。
如果要测二氯乙酰氯残留的话是不是可以直接和正丁醇酯化,然后不中和而是直接顶空进样呢,这样反应生成的盐酸也不会进到色谱柱里吧求大佬们指导一下方法可行性,感谢啊!
项目概况宜春市检验检测中心试剂及耗材和标准品及对照品采购项目(第二次)(第二包:标准物质、对照品) 招标项目的潜在投标人应在 江西省公共资源交易网 获取招标文件,并于 2023年02月09日 09点00分 (北京时间)前递交投标文件。一、项目基本情况:项目编号:明月-YC2022-042-2-1项目名称:宜春市检验检测中心试剂及耗材和标准品及对照品采购项目(第二次)(第二包:标准物质、对照品)采购方式:公开招标预算金额:400000.00 元最高限价:400000.00采购需求:[table=100%][tr][td]采购条目编号[/td][td]采购条目名称[/td][td]数量[/td][td]单位[/td][td]采购预算(人民币)[/td][td]技术需求或服务要求[/td][/tr][tr][td][font=inherit]宜购2022F000814837[/font][/td][td][font=inherit]2022年食品药品监管中央及省级补助资金(标准品、对照品)[/font][/td][td][font=inherit]1[/font][/td][td][font=inherit]批[/font][/td][td][font=inherit]400000.00元[/font][/td][td][font=inherit]详见公告附件[/font][/td][/tr][/table]合同履行期限:详见招标文件本项目不接受联合体投标。
近日在做一个日本上市缓释制剂的仿制,对其释放度检查标准有些疑问,特别是其供试品的吸收度计算方法很特别,希望园里的老师给分析指点一下,谢谢!释放度的测定取本品,照释放度测定法,采用溶出度测定法第二装置,以Ph1.2盐酸溶液500ml为溶剂,转速为每分钟100转,依法操作,经1小时时,取溶液10ml,过滤,作为供试品溶液(1);弃去上述各容器中的酸液,加已预热至37±0.5℃的Ph7.5磷酸盐缓冲液500ml,继续运转至2小时时,取溶液10ml,过滤,作为供试品溶液(2);并补加同体积的Ph7.5磷酸盐缓冲液,继续运转至5小时时,取溶液10ml,过滤,作为供试品溶液(3);另取对照品约0.1g,精密称定,置50ml量瓶中,加甲醇适量使溶解,并稀释至刻度,摇匀;精密量取此溶液各1ml,置100ml量瓶中,分别加Ph1.2的盐酸溶液和Ph7.5磷酸盐缓冲液稀释至此刻度,摇匀,作为对照品溶液(1)和对照品溶液(2)。照分光光度法,供试品溶液(1)在360nm和450nm波长处测定吸收度,计算吸收度差值;其他各对照品溶液及供试品溶液均在360nm波长处测定吸收度,分别计算不同时间的释放量。

最近在做对乙酰氨基酚片溶出度实验,首先测定对乙酰氨基酚的标准曲线,给出的方法如下,仪器为TU1901,对乙酰氨基酚为对照品。[img]http://ng1.17img.cn/bbsfiles/images/2008/05/200805302132_91355_1630080_3.jpg[/img]以上方法是一本教材上的方法,我们严格按照上述方法,溶剂也采用0.04%NaOH,定量测定结果如下,结果很不理想,重配标液后再测,结果同样不理想,后对每一标液做光谱扫描,标1至标4最大吸收波长均在257nm左右,标5和标6最大吸收波长则移动到243nm。 [img]http://ng1.17img.cn/bbsfiles/images/2008/05/200805302137_91356_1630080_3.jpg[/img]经过反复查找资料和分析,推断标5和标6母液取样量大,破坏了溶剂的PH值,致使最大吸收波长发生迁移,遂将溶剂改为0.4%NaOH,重新配制标准溶液进行测定,如果如下图,相关系数很好。 [img]http://ng1.17img.cn/bbsfiles/images/2008/05/200805302150_91359_1630080_3.jpg[/img][color=#DC143C][size=4]总结两点:一、溶液酸碱度对对乙酰氨基酚定量测定有较大影响,实验时应消除PH影响。二、教材及文献的实验方法应动手验证,否则也会有差错。[/size][/color]
近日在做一个日本上市缓释制剂的仿制,对其释放度检查标准有些疑问,特别是其供试品的吸收度计算方法很特别,希望园里的老师给分析指点一下,谢谢!释放度的测定取本品,照释放度测定法,采用溶出度测定法第二装置,以Ph1.2盐酸溶液500ml为溶剂,转速为每分钟100转,依法操作,经1小时时,取溶液10ml,过滤,作为供试品溶液(1);弃去上述各容器中的酸液,加已预热至37±0.5℃的Ph7.5磷酸盐缓冲液500ml,继续运转至2小时时,取溶液10ml,过滤,作为供试品溶液(2);并补加同体积的Ph7.5磷酸盐缓冲液,继续运转至5小时时,取溶液10ml,过滤,作为供试品溶液(3);另取对照品约0.1g,精密称定,置50ml量瓶中,加甲醇适量使溶解,并稀释至刻度,摇匀;精密量取此溶液各1ml,置100ml量瓶中,分别加Ph1.2的盐酸溶液和Ph7.5磷酸盐缓冲液稀释至此刻度,摇匀,作为对照品溶液(1)和对照品溶液(2)。照分光光度法,供试品溶液(1)在360nm和450nm波长处测定吸收度,计算吸收度差值;其他各对照品溶液及供试品溶液均在360nm波长处测定吸收度,分别计算不同时间的释放量。
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
文献里流动相是加了磷酸二氢铵但是实验室没有,可以换成磷酸二氢钠或者磷酸二氢钾么?另外,这三个盐有什么区别啊?谢谢
药典附录中,磷酸缓冲盐不同pH有不同的配制方法,感觉有点繁琐,而且每次配的话,都要拿出药典来对照。我个人觉得只要用一定浓度的磷酸二氢钠和磷酸二氢钠按不同比例混合,再稀释到需要的浓度(用pH计测定),或者先配好一定浓度的磷酸二氢钠,再用氢氧化钠溶液调到相应的pH。药典中配法肯定有他的道理,我想知道的是我的这两种配法各有什么问题或者不足?先谢谢!!!

乙酰基六肽-8,别名阿基瑞林,是一种优质的祛皱化妆品原料, 其抗皱活性高, 副作用小,已在各高端化妆品系列中应用。【详情请咨询国肽生物】它能局部阻断神经传递肌肉收缩讯息,影响皮囊神经传导,使脸部肌肉放松,达到平抚动态纹、静态纹及细纹;有效重新组织胶原弹力,可以增加弹力蛋白的活性,使脸部线条放松,皱纹抚平改善松弛。可用于化妆品内,作为抗皱成分,且效果极佳。产品参数----乙酰基六肽-8/阿基瑞林中文名称:乙酰基六肽-8/阿基瑞林/六胜肽/乙酰六胜肽-3英文名称:Acetyl Hexapeptide-8/Argireline/Acetyl Hexapeptide-3, CAS号:616204-22-9纯度:≥99%分子量 :888.91g/mol分子式 :C34H60N14O12S外观:白色粉末或液体储存条件:2 ℃~8 ℃包装规格(粉末):1g, 10g, 100g包装规格(液体):20ml/瓶,1KG/瓶应用:化妆品原料功效与应用----乙酰基六肽-8/阿基瑞林抗皱抗衰老改善皮肤质量脸部、颈部和手护理品可添加到美容护肤品中,如乳液、面膜、早晚霜、眼部精华液等作用机理----乙酰基六肽-8/阿基瑞林乙酰基六肽-8参与竞争 SNAP - 25 在融泡复合体的位点, 从而影响复合体的形成。当融泡复合体稍有不稳定, 囊泡不能有效释放神经递质, 从而致使肌肉收缩减弱,防止皱纹的形成。[img=,690,143]https://ng1.17img.cn/bbsfiles/images/2020/10/202010141430498557_1196_3531468_3.jpg!w690x143.jpg[/img]国肽生物主要提供:多肽合成、多肽定制、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、美容肽、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。详情请咨询国肽生物
顶空气相色谱法测定缬沙坦原料药中残留溶剂,以二甲基乙酰胺(DMA)作溶剂时,单独的对照不出峰,混合对照出峰,急求各位分析这是为什么?如何改进?顶空条件:DANI HSS86.50 顶空进样器,平衡温度100度,进样系统温度120度,传递管路温度120度,加压时间30秒,压力平衡时间5秒,注入时间30秒气相色谱条件:进样口温度200,检测器(FID)温度250,柱温:初始温度40,维持2分钟,以3度/秒升至100,再以30度/秒升温至200,维持2分钟;分流比10:1色谱柱:rtx-5ms,30m,0.25,0.251、以同样的样品(DMA溶解)手动进样出峰,但顶空不出峰。重复前期品种的方法,配置以水溶剂的甲醇、乙酸乙酯混合对照溶液,顶空进样,结果出峰,这是不是说明问题与DMA有关?2、DMA沸点166度,有没有可能是因为它在系统中冷凝?个人将顶空进样系统温度升至180度后,开始几针样品好了(但仍不确定是不是因为温度的原因),但过了一会单独的对照又不出峰了?3、因为中国药典标准中是用二甲基乙酰胺,所以用它了请各位帮忙分析一下,急死了!有什么没有说明白的地方,请指出,非常非常感谢!
仪器:LC-100 二元高压,Exformma 经济型C18色谱柱,5µm,4.6×250 mm,加保护柱。流动相:乙腈-O.05mol/L磷酸二氢钠溶液(50:50);波长为440 nm;柱温40℃,进样量20μl。理论板数按血竭素峰计算应不低于4000。对照品溶液的制备 取血竭素高氯酸盐对照品9mg,精密称定,置50ml棕色量瓶中,加3%磷酸甲醇溶液使溶解,并稀释至刻度,摇匀,精密量取1ml,置5ml棕色量瓶中,加甲醇至刻度,摇匀.即得(血竭素重量=血竭素高氯酸盐重量/1.377)。供试品溶液的制备取本品适量,研细,取O.05g,精密称定,置具塞试管中,精密加入3%磷酸甲醇溶液10ml,密塞。振摇3分钟,滤过,精密量取续滤液1ml,置5ml棕色量瓶中,加甲醇至刻度.摇匀,即得。血竭素的保留时间为6.4,但拖尾因子为3 ,不符合中国药典规定,请教大家如何处理?
对照品4个,第2(延胡索乙素)、3(盐酸小檗碱)个色谱峰前出现了小峰,不知道什么原因引起的,之前用相同色谱条件跑过,也没有小峰出现,现在连续进了五针都是主峰前有小峰。请各位帮我分析分析!流动相是0.1%的磷酸水:乙腈,对照品用甲醇溶解。色谱条件如图[img=,690,284]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044560750_5451_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044563666_9373_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044551717_7304_5351399_3.png[/img][img=,690,618]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044568830_7416_5351399_3.png[/img]

[size=4][b] 小卢推荐:一种标定二级对照品的方法[/b][/size]对照品作为实验室(制药行业)一种常用的、重要的试剂,根据其类型,可分为:一级对照品,即为从中国药品生物制品检定所(简称:中检所)购买后直接使用的对照品;二级对照品,由一级对照品标定原料药得到的对照品。由于一级对照品的规格小、价格高、购买周期长的缺点,对于实验室对照品用量大的企业来说,使用二级对照品成了实验室的首选。现在,我就介绍一种标定二级对照品的方法,供大家参考一下。[b]第一,选定样品[/b]一般来说,选择自己生产的原料药价格便宜,不需要外购,且取用方便,是我们的首选。如果我们的生产工艺不好、稳定相差,最好选择外购知名企业的原料药。但要注意,要选择作为对照品的原料药一定是相对其他批次各检验项目都比较好的同一批原料药。[b]第二,标定方法[/b]现以高效液相测定法检测含量为例,来表述其测定方法。由于要严格保证所标定原料药的含量,因此采用3人、3份样品的方法进行测定,即:每个人称取2份对照品、3份样品进行测定;共有3人进行测定。如果有条件,3个人可以选择3台不同的液相色谱仪进行实验。在这里要求2份对照品共进样5针,计算校正因子,并求RSD应小于0.5%,3份样品各进1针,求平均值。方法和要求如下表:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182733_1622024_3.jpg[/img]按照表格内容,由3人得到的3个不同的含量,最后求得均值,即得样品(二级对照品)的含量,并要求3者的RSD≤1%。[b]第三,分装[/b]使用抗生素瓶分装,装量按照每次使用量(如60mg,则装入80-100mg即可)为标准,即使每个抗生素瓶中的对照品只使用一次。这样既能避免对照品被污染,又能使其少吸潮。如果为了节省抗生素瓶,采用大装量,即一瓶中的对照品可以使用多次,那么,建议在使用3-6次后就报废本瓶对照品。因为每次打开瓶口称取对照品都是对该瓶对照品的一次污染,尤其是空气中水分对它的影响,这样会是对照品的含量发生变化,原来的标定也就失去了意义。分装环境:建议在层流罩下进行,严格控制温湿度(建议温湿度:18-24℃,45-65%)。封口步骤:分装后,用橡胶盖盖紧,再用封口膜封好后,用铝盖压实即可。[b]第四,制定有效期[/b]一般比较稳定的样品制定2年,不是很稳定的样品制定1年。但是这个有效期不能超过该样品本身法定的有效期。[b]第五,贴签[/b]制定好了有效期就可以把样品(二级对照品)的标签贴上去了,标签格式如下:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182734_1622024_3.jpg[/img][b]第六,储存[/b]不管原来样品法定的存储温度是多少,都建议保存的温度最好在2-10°,即冰箱中的冷藏温度。根据资料研究,2°是药品的最佳保存温度,因为这个温度下药品的降解速度最慢。[b]第七,复核[/b]我们制定了有效期后,并不是就完成了所有的工作。我们要在有效期的一半时,对二级对照品进行复核,检验方法同本法中第二步骤,所取样品则是从原标定的二级对照品中抽取。如果复核结果没有变化,则继续使用;如果复核结果发生了变化,那就按照复核的含量,从新贴签标示。通过以上7步就完成了对照品的标定工作,大家有什么看法可以回帖说明,我们共同讨论![em09505][em09505](全文完!)
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7691601521 二类残留溶剂-1,4-二氧;六环 Residual Solvent Class 2 - 1,4-Dioxane 对照品/标准品1601500 二类残留溶剂-N,N-二甲基酰胺 Residual Solvent Class 2 - N,N-Dimethylformamide 对照品/标准品1601485 二类残留溶剂-N,N-二甲基乙酰胺 Residual Solvent Class 2 - N,N-Dimethylacetamide 对照品/标准品1601463 二类残留溶剂-1,2-二甲氧基乙烷 Residual Solvent Class 2 - 1,2-Dimethoxyethane 对照品/标准品1601441 二类残留溶剂-二氯甲烷 Residual Solvent Class 2 -Methylene Chloride 对照品/标准品1601420 二类残留溶剂- 1,2- 二氯乙烯 Residual Solvent Class 2 - 1,2-Dichloroethene 对照品/标准品1601408 二类残留溶剂-环己烷 Residual Solvent Class 2 - Cyclohexane 对照品/标准品1601383 二类残留溶剂-氯仿 Residual Solvent Class 2 -Chloroform 对照品/标准品1601361 二类残留溶剂-氯苯 Residual Solvent Class 2 - Chlorobenzene 对照品/标准品1601340 二类残留溶剂-乙腈 Residual Solvent Class 2 -Acetonitrile 对照品/标准品1601306 二类残留溶剂-混合物 C Residual Solvent Class 2 - Mixture C 对照品/标准品1601292 二类残留溶剂-混合物 B Residual Solvents Class 2 - Mixture B 对照品/标准品1601281 二类残留溶剂-混合物 A Residual Solvents Class 2 Mixture A 对照品/标准品1601226 一类残留溶剂- 1,1,1- 三氯乙烷 Residual Solvent Class 1 -1,1,1 对照品/标准品1601204 一类残留溶剂- 1,1- 二氯乙烯 Residual Solvent Class 1 -1,1-Dichlo 对照品/标准品1601180 一类残留溶剂- 1,2- 二氯乙烷 Residual Solvent Class 1 -1,2-Dichlo 对照品/标准品1601168 一类残留溶剂-四氯化碳 Residual Solvent Class 1 -Carbon Tetrachloride 对照品/标准品1601146 一类残留溶剂-甲苯 Residual Solvent Class 1- Benzene 对照品/标准品1601102 一类残留溶剂混合物 Residual Solvents Mixture Class 对照品/标准品1601000 利血平 Reserpine 对照品/标准品1600846 瑞格列奈杂质C Repaglinide Related Compound C 对照品/标准品1600835 瑞格列奈杂质B Repaglinide Related Compound B 对照品/标准品1600824 瑞格列奈杂质A Repaglinide Related Compound A 对照品/标准品1600813 瑞格列奈 Repaglinide 对照品/标准品1600121 瑞鲍迪甙 A Rebaudioside A 对照品/标准品1599500 红车轴草提取粉 Powdered Red Clover Extract 对照品/标准品1599000 萝芙碱 Rauwolfia Serpentina 对照品/标准品1598802 树莓酒 Raspberry Alcohol 对照品/标准品1598700 雷尼替丁杂质C Ranitidine Related Compound C 对照品/标准品1598609 雷尼替丁杂质B Ranitidine Related Compound B 对照品/标准品1598507 雷尼替丁杂质A Ranitidine Related Compound A 对照品/标准品1598450 雷尼替丁分离度用混合物 Ranitidine Resolution Mixture 对照品/标准品1598405 盐酸雷尼替丁 Ranitidine Hydrochloride 对照品/标准品1598347 雷米普利杂质D (二酮哌嗪雷米普利)Ramipril Related Compound D 对照品/标准品1598338 雷米普利杂质C Ramipril Related Compound C 对照品/标准品1598323 雷米普利杂质B Ramipril Related Compound B 对照品/标准品1598314 雷米普利杂质A Ramipril Related Compound A 对照品/标准品1598303 雷米普利 Ramipril 对照品/标准品1598201 盐酸雷洛昔芬 Raloxifene Hydrochloride 对照品/标准品1598008 3- 奎宁环基 3-Quinuclidinyl Benzilate 对照品/标准品1597504 奎宁酮 Quininone 对照品/标准品1597005 硫酸奎宁 Quinine Sulfate 对照品/标准品1596807 二水合盐酸奎宁 Quinine Hydrochloride Dihydrate 对照品/标准品1595509 硫酸奎尼丁 Quinidine Sulfate 对照品/标准品1595000 葡萄糖酸奎尼丁 Quinidine Gluconate 对照品/标准品1594506 金鸡纳酸 Quinic Acid 对照品/标准品1594007 喹乙宗 Quinethazone 对照品/标准品1593423 喹那普利杂质 B Quinapril Related Compound B 对照品/标准品1593412 喹那普利杂质 A Quinapril Related Compound A 对照品/标准品1593401 盐酸喹那普利 Quinapril Hydrochloride 对照品/标准品1593004 盐酸米帕林 Quinacrine Hydrochloride 对照品/标准品1592409 槲皮素 Quercetin 对照品/标准品1592227 夸西泮杂质 A Quazepam Related Compound A 对照品/标准品1592205 夸西泮CIV Quazepam CIV 对照品/标准品1592001 恩波吡维铵 Pyrvinium Pamoate 对照品/标准品1589109 丙酮酸 Pyruvic Acid 对照品/标准品1589007 乙胺嘧啶 Pyrimethamine 对照品/标准品1588004 马来酸吡拉明 Pyrilamine Maleate 对照品/标准品
由于手上没有2,3,4-三甲氧基乙酰苯(2,3,4,-trimethoxyacetophenone,C11H14O4)的对照品,所以想求助大家有没有它的光谱图,有的请发一份上来好吗?谢谢大家帮忙啦,
我用液相色谱仪测土霉素原料的杂质时,按照2010年版兽药典一部的规定配置的流动相及相关样品和对照品,土霉素主峰按规定是12分钟出来,可是却4分钟就出来了,且和相邻的2-乙酰-2-去酰胺土霉素未完全分离开,两峰相连的部分在基线上方,柱温25度,这样按外标法计算峰面积时,2-乙酰-2去酰胺土霉素的峰的比例就偏大,超出杂质范围例如,且土霉素峰含量降低了。之后又将柱温设为40度,依旧没有多大改善,如何将两个峰完全分离开且延长出峰时间?(注:两峰相对保留时间约为1.1,这个是正确的)。流动相醋酸铵溶液【0.25mol/L醋酸铵溶液:0.05mol/L EDTA二钠溶液:三乙胺(100:10:1),用醋酸调节PH值至7.5,】:乙腈=88:12

水蒸气蒸馏-乙酰丙酮分光光度法测“血豆腐”中甲醛残留含量前言:“血豆腐”一般是指由猪、牛、羊、鸡、鸭等禽畜动物血做成的凝固状血块,形似豆腐而得名。“血豆腐”向来以营养丰富、口感鲜嫩深得大众青睐,涮火锅、麻辣烫、做汤都少不了它。但有些不法商人为了牟取私利,违法在添加甲醛,甲醛可以让禽兽动物血中蛋白质快速变性凝结,具有防腐作用,还可以使血豆腐表面颜色更漂亮。而甲醛对人体具有很大的伤害作用,对人的肝脏和肾脏损害最大,并具有潜在的致癌性,在食品中严禁使用。为了解市场上“血豆腐”甲醛的残留情况,我们对市场上的“血豆腐”进行了采样监测。方法:参照《卫法监发159号附件2》中测定甲醛次硫酸氢钠的方法原理:根据甲醛沸点低的特点,在酸性条件下对检样进行水蒸汽蒸馏,用水吸收,甲醛馏出后再与乙酰丙酮作用,生成黄色的二乙酰基二氢卢剔啶,依颜色深浅比色定量。仪器与试剂: 普析T6分光光度计; 10%(V/V)磷酸溶液;液体石蜡;乙酰丙酮溶液;甲醛标准溶液(预先标定好浓度)操作步骤:(1)样品处理:称取经均质样品5.00g,置于蒸馏瓶中,加入蒸馏水20ml,液体石蜡2.5ml和10%磷酸溶液10ml,立即通水蒸汽蒸馏。冷凝管下口应事先插入盛有10ml蒸馏水且置于冰浴的容器中,准确收集蒸馏液至于100ml。另做空白蒸馏。(2)显色操作:视检品中甲醛含量高低,吸取检品蒸馏液2-10ml补充蒸馏水至10ml,加入乙酰丙酮溶液1ml混匀,置沸水中浴3-10分钟,取出冷却。然后以蒸馏水调零,于波长435nm处,以1cm比色杯进行比色,记录吸光度。查标准曲线计算结果。(3)标准曲线制备:吸取5ug/L甲醛标准液0、0.50、1.00、3.00、5.00和7.00ml,补充蒸馏水至10ml,以下从加乙酰丙酮溶液起同样操作.减去0管吸光度后,绘制标准曲线。血豆腐样品http://ng1.17img.cn/bbsfiles/images/2014/12/201412251030_528841_2694188_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412251032_528842_2694188_3.jpg取样http://ng1.17img.cn/bbsfiles/images/2014/12/20
因为刚接触药品检验,在做核黄素磷酸钠的游离磷酸的测定的时候,做了几次都不合格。 按照标准要求进行对照及样品配制,对照品为蓝色的澄清液体,但是样品很浑浊,土黄色。在730nm处进行测定,结果不符合规定。 因为样品很浑浊,所以吸收值很大(2.0),大过了对照品; 我们把样品用0.45的滤膜过滤,吸收值(0.35)低于对照,但是我们怀疑过滤的影响太大,我们就再次过滤样品,吸收值又降低了一半(0.13). 请各位大侠帮帮忙啊!!!
食品中甲胺磷和乙酰甲胺磷农药残留量的测定方法1.适用范围本方法适用于谷物、蔬菜和植物油中甲胺磷和乙酰甲胺磷的残留量分析,其最小检出限分别为7.79×10-12g和1.79×10-11g。2.原理概要含有机磷的样品在富氢焰上燃烧,以HPO碎片的形式,放射出波长526nm的特征光,这种特征光通过滤光片选择后,由光电倍增管接收,转换成电信号,经微电流放大器放大后,被记录下来,样品的峰高与标准品的峰高相比,计算出样品相当的含量。3.主要试剂和仪器3.1.主要试剂丙酮;二氯甲烷:重蒸;无水硫酸钠;活性炭:用3mol/L盐酸浸泡过夜,抽滤,用水洗至中性,在120℃下烘干备用;甲胺磷(methamidophos):≥99%;乙酰甲胺磷(acephate):≥99%;甲胺磷和乙酰甲胺磷标准溶液的配制:分别准确称取甲胺磷和乙酰甲胺磷的标准品,用丙酮分别制成0.1mg/mL的标准储备液。使用时用丙酮稀释配制成单一品种的标准使用液(1mg/mL)和混合标准工作液(每个品种浓度为1mg/mL)。贮藏于冰箱中。3.2.仪器气相色谱仪:具有火焰光度检测器;电动振荡器;K-D浓缩器或旋转蒸发器;离心机。4.试样的制备取谷物实验样品经粉碎机粉碎,过20目筛后,制成谷物试样。取蔬菜实验样品洗净,晾干,去掉非食部分后剁碎或经组织捣碎机捣碎,制成蔬菜试样。5.过程简述5.1.提取和净化蔬菜:称取蔬菜试样10g,精确至0.001g,用无水硫酸钠(因蔬菜含水量不同而加入量不同,约50~80g)研磨呈干粉状,倒入具塞锥形瓶中,加入0.2~0.4g活性炭(根据蔬菜色素含量)及80mL丙酮,振摇0.5h,抽滤,滤液浓缩定容至5mL,待气相色谱分析。谷物:称取谷物试样10g,精确至0.001g,置于具塞锥形瓶中,加入40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。小麦:称取小麦试样10g,精确至0.001g,置于具塞锥形瓶中,加入0.2g活性炭及40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。植物油:称取植物油试样5g,用45mL丙酮分次洗入50mL的离心管内,加入5mL水,混匀,在3 000r/min下离心5min,吸取上清液,下面油层再加10mL水和10mL丙酮,离心5min,吸取上清液,合并两次上清液,用K-D浓缩器浓缩近干,残渣和水加入40g无水硫酸钠,研磨呈干粉状,倒入具塞锥形瓶中,加入0.3g活性炭、60mL二氯甲烷,振荡0.5h,抽滤,定容至5mL,待气相色谱分析。5.2.色谱条件色谱柱:玻璃柱,内径3mm,长0.5m,内装2%dEGS/Chromosorb W AWdMCS,80~100mesh。气流:载气,氮气70mL/min,空气0.7kg/cm2,氢气1.2kg/cm2。温度:进样口200℃,柱温180℃。5.3.测定定性:以甲胺磷和乙酰甲胺磷农药标样的保留时间定性。定量:用外标法定量,以甲胺磷和乙酰甲胺磷农药已知浓度的标准样品溶液作外标物,按峰高定量。6.结果计算Xi=hi•Esi•V1hsi•V2•m式中:Xi——样品中i组分有机磷含量,mg/kg;Esi——注入标样中i组分有机磷的含量,ng;hi——样品的峰高,mm;hsi——标样中i组分的峰高,mm;V1——浓缩定容体积,mL;V2——注入色谱样品的体积,μL;m——样品的质量,g。7.方法的精密度添加回收试验中甲胺磷和乙酰甲胺磷的变异系数分别为2.36%和3.95%。8.甲胺磷和乙酰甲胺磷的保留时间在5.2的气相色谱条件下,甲胺磷的保留时间为0.9min,乙酰甲胺磷的保留时间为1.9min。9.来源:GB 14876—94
磷酸三(二甲苯)酯的标品哪里有卖的?CAS:25155-23-1
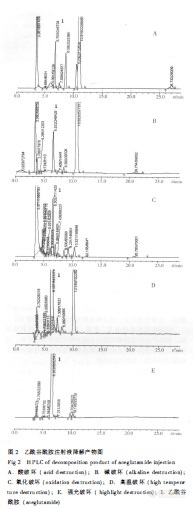
作者:肖菁; 田洪; 梁建国;(湖南省药品检验所;)摘要:目的建立反相高效液相色谱法测定乙酰谷酰胺注射液的有关物质。方法采用Diamonsil C18(4.6 mm×250 mm,5μm)色谱柱,流动相:0.05 mol.L-1磷酸二氢钾(以10%磷酸调节pH至3.0)-甲醇(95∶5),检测波长:210 nm,流速:0.8 mL.min-1,柱温:25℃。结果乙酰谷酰胺与有关物质可完全分离。结论该方法操作简便、快速、结果准确,可用于该制剂的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061059_381740_1606903_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP