GBT 24800.3-2009 化妆品中螺内酯、过氧苯甲酰和维甲酸的测定 高效液相色谱法
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]测定二硫代氨基甲酸酯,标准品不出峰?
http://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471106_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471108_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471109_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152016_471110_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152016_471111_2803904_3.jpg为什么标准品浓度不一样?峰型也不一样? 峰拖尾严重如何处理?标准品怎么会有两个峰?我的用的是C18拄,色谱条件是甲醇:水(0.1%甲酸,10mm的乙酸铵90:10,等度洗脱。做所得是2种磷脂分子,分子式如上
各位朋友,谁有《危险废物鉴别标准 急性毒性初筛》、《危险废物鉴别标准 腐蚀性鉴别》、《危险废物鉴别标准 浸出毒性鉴别》2007年发布的新版,请分享一下,非常感谢!!!
为规范化妆品中禁限用物质检测技术要求,提高化妆品质量安全,化妆品中马来酸二乙酯等禁限用物质的检测方法已经化妆品标准专家委员会审议通过,现予印发 附件:1.化妆品中马来酸二乙酯的检测方法 2.化妆品中环氧乙烷和甲基环氧乙烷的检测方法 3.化妆品中10种着色剂的检测方法 4.化妆品中7种发用品着色剂的检测方法 5.化妆品中诺氟沙星等10种喹诺酮类禁用物质的检测方法 6.化妆品中灰黄霉素等9种抗真菌类禁用物质的检测方法 7.化妆品中α-氯甲苯的检测方法 8.化妆品中禁用物质氨基己酸的检测方法 9.化妆品中氯苯甘醚的检测方法 10.化妆品中乙醇胺等5种有机胺的检测方法 11.化妆品中维甲酸和异维甲酸检测方法 12.化妆品中山梨酸和脱氢乙酸的检测方法 13.化妆品原料丙二醇中二甘醇检测方法 14.化妆品中吡硫翁锌等5种物质的检测方法
在配置邻苯二甲酸脂的标准品的过程中,用正己烷配置的一定浓度标准品比如1.0ppm 在常温下放置一周,体积并无变化.为何上机分析时与一起配置的相同浓度(1.0ppm存储于冰箱中)峰面积相差几倍?(检测仪器重复性很好)
本人正在做中药的生物碱提取,我之前看了资料,对样品进行了初步提取(最后一步是氯仿萃取),然后选择改良碘化铋钾、碘-碘化钾和磷钼酸进行定性,但是当我把这三种试剂分别滴到样品之后,问题来了:1、 理论说这三种试剂如果有阳性反应的话,是呈橘红色、棕黄色的,但我滴下去后,出现的是分层了,而没有沉淀出现。为什么大家都说加进去后会有沉淀呢?样品的溶剂是氯仿,而沉淀试剂的溶剂是稀冰醋酸,这两者是不相溶的,那何来的反应出现沉淀呢?2、我要提取的生物碱,目前还没有标准品可以购买,对于这种没有标准品的生物碱,要拿什么做对照品呢?请大家多多指教。我查了文献,也有其他人在做同样这种情况的实验(没有标准品的生物碱提取),但他们是用其他的生物碱作为对照品来鉴别的,可以这样的吗?这样做的依据是什么呢?好迷茫啊,希望大家多多回复啊!!!!谢谢了!
本人正在做中药的生物碱提取,我之前看了资料,对样品进行了初步提取(最后一步是氯仿萃取),然后选择改良碘化铋钾、碘-碘化钾和磷钼酸进行定性,但是当我把这三种试剂分别滴到样品之后,问题来了:1、 理论说这三种试剂如果有阳性反应的话,是呈橘红色、棕黄色的,但我滴下去后,出现的是分层了,而没有沉淀出现。为什么大家都说加进去后会有沉淀呢?样品的溶剂是氯仿,而沉淀试剂的溶剂是稀冰醋酸,这两者是不相溶的,那何来的反应出现沉淀呢?2、我要提取的生物碱,目前还没有标准品可以购买,对于这种没有标准品的生物碱,要拿什么做对照品呢?请大家多多指教。我查了文献,也有其他人在做同样这种情况的实验(没有标准品的生物碱提取),但他们是用其他的生物碱作为对照品来鉴别的,可以这样的吗?这样做的依据是什么呢?好迷茫啊,希望大家多多回复啊!!!!谢谢了!
日本岛津液相色谱仪检测食品中苯甲酸含量,流动相是乙酸铵(95%)和甲醇(5%),检测器为PDA,230nm下标样出峰时间是11.711,可是样品出峰时间为12.260,时间相对标准偏差为3.24%,怎么判定是苯甲酸呢?
药品质量标准中鉴别项目设置的几点考虑 审评三部 张哲峰 摘要:本文简要介绍了药品质量标准中常用的几种鉴别方法,并对常用鉴别方法的优势和局限进行了分析,针对鉴别项目设置中需注意之处提出了一些看法。 关键词:质量标准 鉴别项目 药品质量标准中鉴别是用以判定某已知药品的真伪而不是对未知药物进行结构确证,所以鉴别方法应以专属性好、简便易行为宜,尤其能将结构相似的同类药品加以区别为主要考虑因素。如新鱼腥草素钠及制剂标准中仅用化学法和UV法作鉴别,难以与结构类似物鱼腥草素钠及制剂相区分,质量标准不具备应有的专属性,可能给此后的市场监督造成混乱。 常用的鉴别方法包括色谱法、光谱法、化学法和生物学方法等,可根据药品具体特点加以选用。 色谱法(TLC法或HPLC法)利用不同物质在不同色谱条件下,各自色谱行为(比移值或保留时间)的不同,与对照品在相同色谱条件下进行色谱分离,比较其色谱行为的一致性,来鉴别药品的真伪。这类方法的运用使得结构相似化合物、同系物等的区分变得简单易行。HPLC法虽然主要用于定量,但如果运用得当,尤其在含量测定或有关物质项下已采用本法的情况下,利用对照品与供试品保留时间相同的特性作为鉴别依据,不必专门增加实验以提高鉴别的专属性,是非常可取的。值得注意的是色谱系统的稳定性要好,同一物质不同进样时保留时间的重现性必须有保证。这就要求流动相与固定相相匹配,C18链在水相环境中不易保持伸展状态,故在C18柱的反相色谱系统中,流动相有机溶剂比例通常不应低于5%,否则C18链的随机卷曲将造成色谱系统不稳定导致组份保留值波动,不利于此种鉴别。即便如此,在实际操作中有时依然能遇到同一物质在完全相同的色谱系统中保留时间不一致的情况,尤其梯度洗脱时此种现象更为常见。药典中对保留时间的一致性未予具体规定,此时,操作中可增加供试品溶液与对照品溶液等量混合,进样后出现单一色谱峰作为鉴别依据,可以弥补该法之不足,此操作可列入质量标准。在含量和有关物质未采用HPLC法的情况下,一般不单独采用本法作鉴别。 TLC法除色谱行为外,还可将斑点颜色作为鉴别依据,可由两个因素把握供试品与对照品的同一性,而且简便易行,堪称一个很好的鉴别方法。但由于薄层板质量、边缘效应等因素的影响,实际操作中有时也会遇到同一物质在同一块薄层板上的Rf值不一的情况,可比照HPLC的情况,操作中增加供试品溶液与对照品溶液等量混合,点样后出现单一斑点作为鉴别依据,此点在2005年版药典中已有体现。也有人提议明确Rf值偏差不超过5%,作为鉴别要求,但其可行性有待考察。单独使用TLC鉴别时,要有色谱系统适应性试验内容,如要求几种结构相似化合物的混合溶液色谱展开后应显示相应的几个斑点或最难分离物质对能够分开的情况下,供试品溶液与对照品溶液主斑点的颜色与位置应一致。 在中国药典2005年版中,对TLC鉴别法在斑点的颜色与位置明确规定的基础上对斑点大小也做出明确要求:供试品与同浓度对照品溶液颜色与位置应一致,斑点大小应大致相同;或供试品与对照品等体积混合,应显示单一,斑点紧密;或供试品溶液的主斑点与上述混合溶液的主斑点的颜色与位置一致,大小相似;或选用与供试品化学结构相似药物对照品,两者的比移植应不同(例如芬布芬与酮洛芬,地塞米松磷酸钠与泼尼松龙磷酸钠,醋酸氢化可的松与醋酸可的松,泼尼松龙与氢化可的松,甲睾酮与睾酮,左旋多巴与酪氨酸);或上述两种溶液等体积混合,应显示两个清晰分离的斑点。 光谱法中IR法因可反映较多的结构信息,在组份单一、结构明确的原料药鉴别中作为首选, 药物存在多晶型现象又无可重复转晶方法时一般不采用此法,但如果药物存在多晶型现象,且需鉴别其有效晶型,IR图谱可以反映其有效晶型特点时,本法又是一种有效而简便易行的鉴别方法。制剂中则因辅料影响、提取过程可能导致晶型变化而一般不采用IR法,而采用所受影响因素较少的UV法。 常用的UV鉴别方法有:测定最大吸收波长,或同时测定最小吸收波长;规定一定浓度的供试液在特定吸收波长(最大吸收或最小吸收)处的吸收度;经化学处理后,测定其反应产物的吸收光谱特征;规定几个特定吸收波长及其吸收度比值(峰-峰、峰-谷、谷-谷);规定几个特定吸收波长及其吸收系数。因末端吸收所受影响因素较多,UV法鉴别时,一般不宜用220nm以下波长的吸收特性作鉴别;因反映的结构信息少,一般也尽量不用单一吸收峰作鉴别依据;为提高专属性,可将上述几个方法结合起来使用。 化学鉴别法一般是特定官能团或特定结构化合物的特性反应,与其它鉴别方法结合使用,可以使得鉴别的专属性更加突出。化学鉴别法具有专属性较强、反应迅速、现象明显的特点才有使用价值。包括在适当条件下产生颜色、荧光,发生沉淀反应或产生气体等现象。 1.呈色反应:即向供试品溶液中加入适当试剂,在一定条件下发生化学反应,生成易于观测的有色产物。常见的反应类型有:[/c
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]型号是岛津的GC 2010 PRO。色谱柱WondaCap-5(30m*0.25mm*0.25μm)。进样口温度280℃,FID检测器温度300℃,升温程序:初温60℃,保持1min,以20℃/min升至220℃,保持4.5min,以5℃/min升至250℃,保持1min,再以15℃/min升至290,保持5min,流速1.0mL/min,进样量1μL,不分流进样。进样标液的浓度为20mg/L,标准品新拆的。换了进样垫,用的高惰性衬管。有溶剂峰,目标物不出峰。色谱柱期间也试过DB-1701,也是有溶剂峰,目标物邻苯二甲酸二(2-乙基己基)酯不出峰。各位老师,能帮忙看看是什么原因吗。[img]https://simg.instrument.com.cn/bbs/images/default/em09509.gif[/img]

如题,有哪位大神指导一下如何配8种邻苯二甲酸盐的标准品溶液,各浓度如下图,需把八种邻苯二甲酸盐配制混在一个容量瓶中,然后进行上机检测得到标准品图谱。谢谢比如,我买的八种邻苯二甲酸盐的其中一个:DEHP.标准品,是液体的,瓶装的,0.25g。怎么样配制最终浓度为50ppm?http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_668782_3017235_3.jpg
本标准代替GB/T21911—2008《食品中邻苯二甲酸酯的测定》和SN/T3147—2012《出口食品中邻苯二甲酸酯的测定》。 本标准与GB/T21911—2008 相比,主要变化如下: ● 标准名称修改为“食品安全国家标准 食品中邻苯二甲酸酯的测定”; ● 增加了邻苯二甲酸二烯丙酯和邻苯二甲酸二异壬酯两种目标化合物; ● 增加了同位素内标法定量作为第一法。 新国标对应的标准品是17 种混标+1 种DINP 单标的形式: ●E.1 邻苯二甲酸二异壬酯(DINP)标准溶液(1.0μg/mL)的总离子流色谱图(外标法)见图E.1。 http://www.anpel.com.cn/UpFile/Admin/image/20170413/20170413100030_9357.jpg 图 E.1 邻苯二甲酸二异壬酯(DINP)标准溶液(1.0μg/mL)的总离子流色谱图(外标法) ●E.2 17种邻苯二甲酸酯标准溶液(0.12μg/mL)的总离子流色谱图(外标法)见图E.2。 http://www.anpel.com.cn/UpFile/Admin/image/20170413/20170413100208_5899.jpg 图 E.2 17种邻苯二甲酸酯标准溶液(0.12μg/mL)的总离子流色谱图(外标法) DNP 和DINP 的解读: ● Cas 84-76-4 邻苯二甲酸二壬酯(DNP 单峰); ● Cas 28553-12-0 是邻苯二甲酸二异壬酯(DINP)一类同分异构体的混合物,此物质适宜做标准品; ●Cas 68515-48-0 是邻苯二甲酸酯的混合物,含有三类同分异构体: 邻苯二甲酸二异辛酯(DIOP), 邻苯二甲酸二异壬酯(DINP), 邻苯二甲酸二癸酯(DIDP),其中主要成分是DINP。 推荐标准品: http://www.anpel.com.cn/UpFile/Admin/image/20170413/20170413100549_9252.jpg
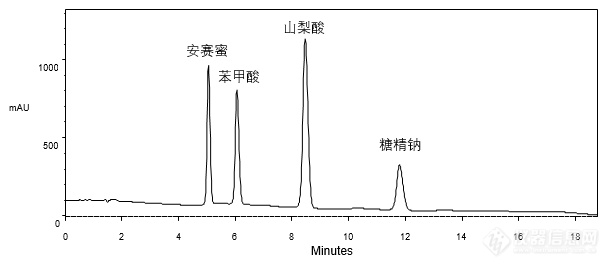
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
各位老师,请教一个GCMS检测邻苯二甲酸酯的问题,检测标准品时,每个峰前都会有一个小峰,采购的100ppm原标是用甲醇配制的,校准曲线是用丙酮稀释的,大家有遇到过这个现象吗?是甲醇丙酮溶剂汽化体积不同造成的吗?谢谢。
蜂蜜检测仪是一种专门用于鉴别蜂蜜真假的设备,它基于先进的技术手段,能够准确、快速地分析蜂蜜中的成分,从而判断其真伪。 蜂蜜检测仪的操作步骤 准备样品:从待检测的蜂蜜中取出适量样品,确保样品具有代表性。同时,准备好标准蜂蜜样品作为对照。 设置检测参数:根据蜂蜜检测仪的说明书,设置合适的检测参数,包括检测波长、测量时间等。这些参数的设置对于测量结果的准确性至关重要。 进行测量:将蜂蜜样品和标准样品分别放入蜂蜜检测仪的检测通道中,启动仪器进行测量。仪器会自动对样品中的蔗糖、果糖、葡萄糖以及甲基糠醛等成分进行分析,并给出相应的数据。 分析测量结果:测量完成后,可以通过仪器的显示屏查看每个样品的测量数据。将待检测蜂蜜的数据与标准蜂蜜的数据进行对比,如果两者差异较大,则很可能是假蜂蜜或者掺假蜂蜜。 蜂蜜检测仪能够准确鉴别蜂蜜的真假,主要体现在以下几个方面: 成分分析:蜂蜜检测仪能够精确测量蜂蜜中蔗糖、果糖、葡萄糖等关键成分的含量,从而判断蜂蜜是否纯正。 掺假检测:通过对比待检测蜂蜜与标准蜂蜜的数据,可以判断蜂蜜是否存在掺假行为,如添加糖精、水等。 快速高效:蜂蜜检测仪能够在短时间内完成大量样品的检测,提高了检测效率。 总之,蜂蜜检测仪是一种有效的工具,能够帮助我们准确鉴别蜂蜜的真假。通过科学、规范的操作和维护,可以充分发挥其鉴别能力,保障消费者的权益。 在农产品收购环节,可以使用这些设备进行快速筛查,避免收购到农药残留超标的农产品。在食品加工前,对原料进行农药残留检测,确保原料的安全性,防止因原料问题导致的食品安全事故。 在食品加工前,对原料进行农药残留检测,确保原料的安全性,防止因原料问题导致的食品安全事故。 在销售前对农产品进行快速检测,确保销售的农产品符合食品安全标准,保障消费者的健康。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/07/202407081453095005_4648_6238082_3.jpg!w690x690.jpg[/img]
有药品/食品用复合塑料袋的红外鉴别标准图谱吗?
各位大神,怎么配制邻苯二甲酸盐标准品?我想配个8p,就是把八种邻苯二甲酸盐的标准品混在一个容量瓶,
《食品中氨基甲酸乙酯测定》国家食品安全标准终于制定完成发布,大家在使用过程有什么问题,可以联系我互相交流。
食品安全质量鉴别与国家检验标准全书[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=114963]食品安全质量鉴别与国家检验标准全书[/url][img]http://ng1.17img.cn/bbsfiles/images/2008/10/200810271936_114964_1634467_3.jpg[/img]
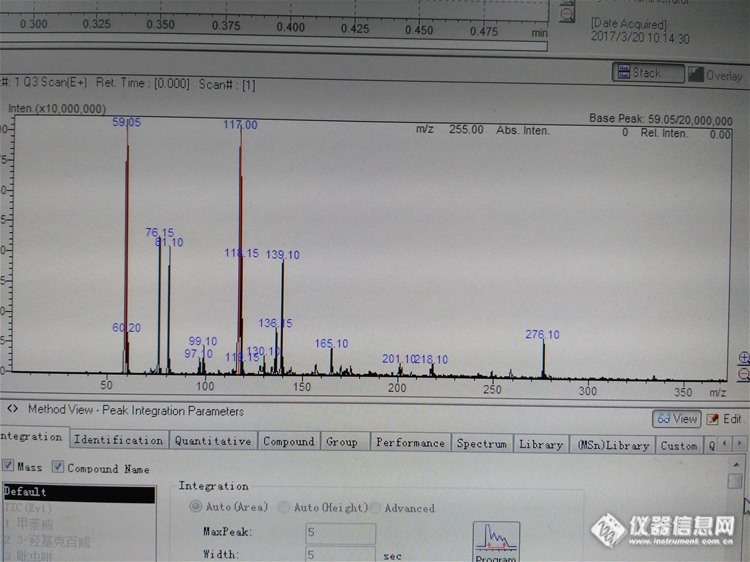
http://ng1.17img.cn/bbsfiles/images/2017/03/201703201613_01_2725311_3.jpg如题,配的1ppm的几个标准品杂峰怎么这么高,目标物都被淹没了,如果是污染了,大概是系统什么地方污染了?什么物质污染的?标准品:甲萘威 等,注射进样,1uL,Q3Scan,流动相0.05%甲酸水+乙腈
[color=#444444]安捷伦1200[/color][color=#444444] 流动相:乙腈:1%甲酸水溶液=20:80(v/v); 流速:1ml/min;柱温30度;检测波长:280nm[/color][color=#444444]杂峰出现在物质峰之前,不干扰物质峰;杂质峰面积随标准品浓度的增加有上升趋势,但非线性;物质峰线性很好R2达0.999以上;标准品:流动相溶解&定容;以流动相为空白无此峰(面积极小)[/color][color=#444444]两种喹诺酮类抗生素标准品皆出现此状况;求指点[/color]
危险废物鉴别标准 浸出毒性鉴别 GB 5085.3-2007 4.4中规定:“各危害成分项目的分析,除执行规定的标准分析方法外,暂按附录中规定的方法执行;待适用于测定特定危害成分项目的国家环境保护标准发布后,按标准的规定执行”。请问“其中铜、镍的测定是否可以执行《[font='Times New Roman'][font=宋体]固体废物[/font] [font=宋体]镍和铜的测定[/font] [font=宋体]火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/font]HJ[/font][font=宋体] [/font][font='Times New Roman']751-2015[/font]》或《[font='Times New Roman'][font=宋体]固体废物[/font] [font=宋体]铍[/font] [font=宋体]镍[/font] [font=宋体]铜和钼的测定[/font] [font=宋体]石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/font][/font][font='Times New Roman']HJ[/font][font=宋体] [/font][font='Times New Roman']752-2015[/font]》”?
国标FZ_T 01057-2007(2-9部分介绍了纤维鉴别方法)ISO 1833:2006 (2-24部分介绍了纤维定量方法)ISO 1833-1-2006 测试总则(谁有中文版http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif)ISO标准中 是否有详细的纤维鉴别方法的标准???http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif和国标的是否有什么区别?http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif
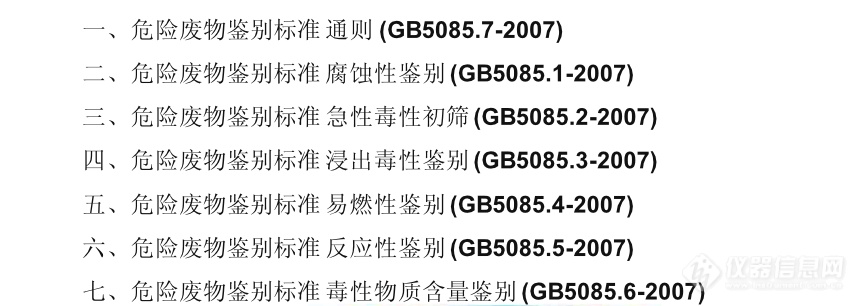
国家环保部与国家质量监督检验检疫总局联合发布危险特性鉴别标准名称、编号如下:[img=,690,245]https://ng1.17img.cn/bbsfiles/images/2022/07/202207281156386533_7307_3141805_3.png!w690x245.jpg[/img]
[color=#444444]我利用色谱分离得到一定量的化合物标准品,纯度用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]检测后纯度比较高,但是里面可能含有检测不出来的甲酸铵,由于流动相中含有的电解质。我们用凝胶柱分离后不知道甲酸铵是否可以除干净。。。请各位大侠帮忙指点如何去除甲酸铵以及用过凝胶柱的方法是否可以将甲酸铵除尽???我的化合物分子量300不到,水溶性比较好,纯的化合物貌似是油状的。。。多谢各位帮忙指点迷津!!![/color]

GC-MS测定食品中邻苯二甲酸酯的不确定度评定1. 测试方法简述1.1 测试步骤测量不确定度是表征合理赋予被测量之值的分散性,与测量结果相关联的参数,其大小决定了测量结果的使用价值。本实验采用气相色谱-质谱联用仪(GC-MS),对食品中邻苯二甲酸酯质量浓度进行分析检测。我们基于JJF1059-1999《测量不确定度评定与表示》的一般要求,分析测试过程中不确定度的主要来源,评估标准不确定度、合成不确定度和扩展不确定度的数值,通过不确定度的分析结果来评定该方法的适用性。本报告主要以测试食品中的邻苯二甲酸二(2-乙基己基)酯(DEHP)为例子,其他分析测试过程相同。1.2 实验原理和步骤1.2.1 实验原理本实验依据GB/T 21911-2008的标准方法,进行萃取、鉴别和量化食品中含有的邻苯二甲酸酯。以正己烷作为萃取溶剂,采用气相色谱-质谱联用仪(GC-MS),鉴别并量化各种邻苯二甲酸酯。1.2.2 设备和试剂1.2.2.1正己烷(AR);邻苯二甲酸酯的标准溶液(DBP、BBP、DEHP、DNOP、DINP、DIDP 美国Dr.E)1.2.2.2 梅特勒SHSB071分析天平(±[size=10.
[color=red]【由于该附件或图片违规,已被版主删除】[/color][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=54454]GB11311-91品容器及包装材料用聚对苯二甲酸乙二醇酯成型品卫生标准[/url]
配制邻苯二甲酸酯类标准品时,购买的标准品是纯品,但是也是液体的,100mg,配制的时候,一次全部清洗到100ml棕色容量瓶中,标准品溶液的浓度是多少?是1mg/ml吗?还是要用减量法称量一下具体的含量。谢谢!我用正己烷溶解定容的,保存的时候直接放在-10°的冰箱冷冻室保存,可以吗?说什么的都有,如果是称量的话,这样也不好吧,做邻苯二甲酸盐的来说说吧
对羟基苯甲酸甲乙丙酯的标准溶液有很多杂峰正常吗?我感觉理论上应该是只有三个大的色谱峰,结果出了十多个[img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310191022364344_1417_5979722_3.png[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP