各位同仁:新购进了固体二氯二甲吡啶酚标准品,请各位指教用什么试剂溶解比较好?谢谢!
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
吡啶二羧酸酐结构看似简单,可因其在水中水解成酸,影响样品游离酸含量分析,化学滴定不好作;做GC时热分解,与相应的二酸峰重合;液相用水溶液作流动相不行,用非水有机相作流动相如用醇又会醇解,估计可用正相做,但该样在许多有机溶剂中溶解性差,所以一直没找到好的分析方法。请问哪位老师作过吡啶二羧酸酐的HPLC分析,能否帮帮我。谢放!
本人现急需氯亚铂酸钾和二(乙酰丙酮)铂的分析方法或行业标准,那位大虾有相关资料,请上传或发到我的信箱[color=#DC143C]zhangfy03@126.com[/color],不胜感激!!!!
据欧盟食品安全局(EFSA)消息,依据欧盟委员会(EC)No396/2005法规第6章的规定,芬兰收到一份申请要求修定部分作物中农药二氯吡啶酸(clopyralid)最大残留限量的申请,为协调欧盟北部地区与南部地区二氯吡啶酸的残留限量,芬兰建议提高该农药在部分作物以及部分动物性食品中的最大残留限量。 芬兰依据欧盟委员会(EC)No 396/2005法规第8章的规定对此起草了一份评估报告,并提交至欧委会,之后于2011年3月8日转至欧盟食品安全局。 欧盟食品安全局对评估材料进行审核后,做出如下决定: 商品代码 商品现行MRL(mg/kg)建议MRL(mg/kg) 241020菜花0.53.0241010西兰花0.51.5242020甘蓝0.5 3.0401010亚麻籽1.02.0213100瑞典芜菁0.51.5213110芜菁0.51.51012010牛肉0.050.081013010绵羊肉1014010山羊肉1012030牛肝0.050.061013030绵羊肝1014030山羊肝1012040牛肾0.050.41013040绵羊肾1014040山羊肾1020000牛奶0.050.015
GB 29699-2013 食品安全国家标准 鸡肌肉组织中氯羟吡啶残留量的测定 气相色谱-质谱法
GB 29700-2013 食品安全国家标准 牛奶中氯羟吡啶残留量的测定 气相色谱-质谱法
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析脂肪酸甲酯标准品,怎样处理标准品?用什么溶剂稀释?

【作者】 方奕珊; 李来生; 陈红; 张杨;【Author】 FANG Yi-shan,LI Lai-sheng,CHEN Hong,ZHANG Yang(The Centre of Analysis and Testing,Nanchang University,Nanchang 330047,China)【机构】 南昌大学分析测试中心;【摘要】 以4-二甲氨吡啶为内标,发展了一种高效液相色谱法,用于快速测定鸡蛋中氯羟吡啶的残留量。鸡蛋样品中的氯羟吡啶经乙腈匀浆提取,然后用碱性氧化铝柱富集,甲醇洗脱液浓缩后用流动相溶解残渣,0.45μm滤膜过滤后进样分析。色谱柱采用Platisil-C18柱(4.6mm×250mm,5μm),流动相为0.05mol.L-1甲酸铵-乙腈(85:15,v/v,pH 6.4),流速为0.8mL.min-1,二极管阵列定量检测波长为267nm,柱温25℃,进样量20μL。氯羟吡啶在0.5~80μg.mL-1范围内呈现良好的线性关系,线性方程为Y=0.254 4 X+0.703 9,相关系数为0.999 6。3个不同水平的标准添加平均回收率分别为85.3%,88.2%和83.7%,相对标准偏差≤2.25%(n=4)。最低检出限为1.8mg.kg-1。该方法快速、简便、准确、重现性好,适用于鸡蛋中兽药氯羟吡啶残留量的快速检测。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301524_380581_2379123_3.jpg
有谁知道哪个单位有99.9%含量的吡啶甲铬标准品出售?必须附带证书。急需!
如题,跪求2-甲基吡啶、2-乙烯基吡啶检测标准,有谁做过没?急求急求急求。。。。。有知道的回复我吧。。。
请问乙二胺四乙酸分析纯能直接配制EDTA标准溶液吗?请如果能配制有方法吗,比如配0.1mol/L的,请各位帮忙,我手里现在只有乙二胺四乙酸分析纯
[color=#333333]食品中磷化铝的标准分析方法[/color]
我们实验室是检测饲料和原料含量的,以前别人介绍用过中国农业科技院分析检测中心研制的氨基酸分析用校核标准品(参比物),名字我忘记了,只记得有代号,1#、2#、3#、4#、5# 这5种。有知道的老师们请帮帮忙!
用高效液相色谱分析:邻氯苯基环戊酮; 2-吡啶甲醛;方酸; 2,5-二氧基-4-碘苯乙胺; 苯瞵酸; 戊亚酮胺的含量的检测条件,恳请各位专家帮帮忙啊!
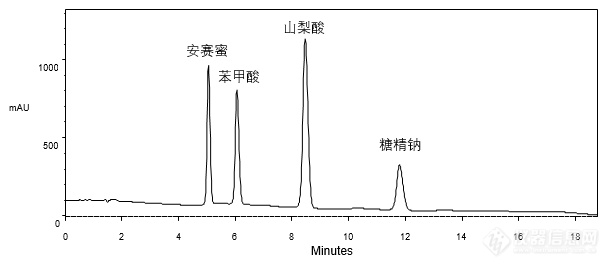
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
食品分析中标准物的管理及其标准溶液的校正一、意义食品分析标准物质是分析方法质控的核心,是定性和定量的依据。标准物质以一定纯度和浓度配制的溶液称标准溶液,其稳定性受其自身降解、化学转化、溶质和溶剂挥发等内在因素的影响,又受其存放条件如温度、湿度、光线照射、存放时间、存放容器及其配制技巧等外界条件的影响。由于影响标准物及其标准液的因素较多,如果条件控制不当或管理不严密,标准物质浓度易发生变化,这是食品分析中难以进行质量控制的主要原因。以上原因也是实验室食品分析测定产生误差的最主要因素,要减小这些产生误差的主要因素,就要特别注重标准物管理,以及进行标准溶液的稳定性观察和校正工作,也是实验室质量控制关键工作之一。当标准溶液发生变化时就要重新配制,并找出变化原因,为分析工作积累经验,并可写入方法注释中。在食品分析质量控制中,准确度和精密度的提高,是以标准溶液稳定性和准确性为前提的,因此对于标准溶液稳定性和准确性的关键技术问题,是质量控制的核心问题。二、标准物质的管理 1、容量分析的基准物是标定其它标准溶液的基准,应购买基准试剂,它可以保证其纯度。另一个因素是水份的影响,用前应充分干燥和恒重,配制时量器要校正。溶剂要纯化,使用的容器要充分洗涤。最终目的都是防止基准物的化合损失。2、用于分光光度分析的标准物,分有机的和无机的标准物,无机标准稳定性好,但使用液浓度低时,极易被容器吸附,并与容器中离子进行交换,因此决定了其稀溶液使用时间短。玻璃容器在碱性介质中易溶出,塑料容器在成型时加入助剂时也含有不同金属杂质,容易溶出如Zn、Ca等金属离子。有机标准最好不放在塑料制容器中,因塑料在成型时加入的有成份比较复杂的助剂和增塑剂。标准使用液应现用现配为最佳。三、标准物存放使用1、无机的标准物要求在干燥并无化学干扰物的条件下存放,选择合适干燥剂如硅胶和分子筛。有机标准物最好分装封入安培瓶中低温避光保存。固体的多环芳烃可配制成溶液,再分装在安瓿瓶中保留溶剂封存,也可把溶剂挥掉后干燥封存,使用时再定容,后一种方法更为稳妥些。配制好一批标准溶液,再一支一支使用也是很方便的。如果液体的标准物特别是几种标准的混合物用于色谱分析同系物如醇类物质,可同时配制一批分装安瓿瓶低温存放,再一支一支使用,能避免溶剂挥发体积变化产生的误差。这种做法更适于实验室间的标准分发和校正工作。最难办的是气体,标准如氯乙烯、氟里昂,最好是钢瓶中存放,或配制钢瓶标准气。这些条件不具备时也可以选择高沸点溶剂,密封溶解这些气体,称量溶质重量,一次性使用。从这一事实出发,气体,测定误差可以稍加放宽,因为标准自身稳定性差。2、用于色谱分析的标准物要求色谱纯,其配剂溶液剂也要求色谱纯,准确配制前要在色谱上进行检查。特别是几种标准进行混合更应慎重,每种要严格检查否则给定性定量带来很多麻烦。如果纯度不够时可以纯化,再结晶或用制备色谱制备。勉强使用是无益的。四、标准溶液的校正1、从安瓿瓶分装标准溶液无论是有机的或是无机的用于校正是很方便的。如原子吸收测定金属,从安瓿瓶中取一定量配制浓度系列,再封存。每隔一段时间(1~2周)再用原溶液配制同样浓度系列,严格控制仪器条件来比较二次标准曲线的斜率,斜率下降时表明有损失。2、相同浓度同时配制的标准的几支安瓿瓶,先用打开的一支标准的测定值与间隔一段时间后打开的另一支标准的测定值进行比较,以此类推最后在一段时间内几支同时测定,其变异程度就是标准在这段时间稳定性变化程度。3、几个实验室用同一标准物分别配制相同浓度标准液,各自进行标准曲线的测定,再按规定交换该标准液再进行测定,比较测定结果差异来观察同一标准的时间和空间变异。如果标准液稳定,配制不准确的实验室很容易查出。配制都准确时,标准液若不稳定时,会使各实验室的测得值都偏低。4、同种标准物来源不同,也应采用分别配制交叉测定的办法来检查标准的纯度及配制是否准确。在食品分析中无论用何种手段分析样品,所使用的标准物应作统一的或确切的规定。例如:过硫酸铵测锰,用MnSO4·H2O作为标准使用,到底硫酸锰需要不需要烘烤呢?对于这个问题,在一部份的教科书中有规定烘烤的,也有不烘烤的。按照MnSO4·H2O的性质遇到空气可能吸潮或风化,如果直接称重计算Mn量,就有可能出现误差。用烘烤称重测得水分所含的量比理论值高1.6%,有同一硫酸锰配制锰标准溶液测一合成水样,使用烘烤后配制的锰标准溶液,测得的Mn含量为0.205mg/L,未烘烤过的则高达约2.3%,从中说明硫酸锰在配制标准溶液时应经过烘烤,使标准一致。
各位同仁!请教你们一个问题,我现在在做3-甲基吡啶和3-氰基吡啶的一个液相分析,目前我把4-个标品买回来了,分别是3-甲基吡啶,4-甲基吡啶,3-氰基吡啶,4-氰基吡啶;目前的一个情况就是3-甲基吡啶和4-甲基吡啶液相无法分开,3-氰基吡啶和4-氰基吡啶无法分开,液相打出来完全重合,我用的柱子是岛津C18柱子。流动相是甲醇:异丙醇:庚烷磺酸钠溶液=7:2:91,流速:1.0ml/min,检测波长261nm,请问有谁做过这样的液相分析,能否告诉小女子一下,万分感谢!
如题,跪求空气中2-甲基吡啶、2-乙烯基吡啶检测标准,有谁做过没?急求急求急求。。。。。有知道的回复我吧。。。
做SPS的分析,但是没有标准品,只好用国外产品做标样。但是老板不认可国外产品含量标注。无法,只好上来求助。SPS:http://www.chemyq.com/xz/xz1/2062nilyd.htm,这个是化工引擎上的介绍。聚二硫二丙烷磺酸钠,结构式 NaSO3(CH2)3-S-S-(CH2)3SO3Na。我目前使用C18柱,uv检测器,50%甲醇加离子对试剂。

研究概况 物质的相平衡数据测定及其相平衡研究是化工热力学的一个重要分支,固液相平衡是化工分离的理论基础。固液平衡的研究为结晶分离过程规定了分离极限,并为设备结构尺寸的设计和操作条件的确定提供基础数据,是实现化工生产的重要前提。实验测定固液平衡不仅是工程设计必不可少的基础数据,也是进行理论研究的基础。固液平衡的数学模型参数需要由实验数据来回归,数学模型的准确性需要用实验数据来检验。通过对固液平衡实验数据的处理,找出其内在规律,提出符合溶解行为的数学模型。 4-二甲氨基吡啶(简称DMAP)结构上有共电子的二甲氨基与吡啶环的共振,强烈的激活了环上的氮原子进行亲核取代,明显催化高位阻、低反应性的醇或胺的酰化(磷酰化、磺酰化)反应,其活性为吡啶的106倍。1967年,Litvinenko和Kirichenko用间氯苯胺进行苯甲酰化动力学研究,以4-二甲氨基吡啶代替吡啶,发现其反应速率增加约1万倍。Steglich、Hassner等人开始着手于研究DMAP作为催化剂催化酰化反应,酰化反应应用于醇、胺、酚和烯醇盐,尤其是存在空间位阻的仲醇、叔醇等。至今,DMAP在酰化反应催化剂中,具有反应速度快、副反应少、溶剂选择范围广、反应条件温和、反应温度低、催化剂量少、对空间位阻大与活性低的醇类酰化催化效果明显的特点,被称为“超级催化剂”。 美、欧、日等国家早已实现DMAP的工业化生产与应用研究,广泛地应用于医药、高分子、精细化工等行业中,我国从90年代初开始DMAP的合成与应用研究,目前,在化学制药领域上取得了成功的应用及良好的效益,如乙(丙)酞螺旋霉素、青篙素唬拍酸酷等原料药的生产;农药领域上在胺菊酯的合成已通过中试并投产。1.实验部分1.1实验试剂4-二甲氨基吡啶 (1) DMAP的物性:白色结晶性粉末,溶于水、乙醇、丙酮、苯、甲苯、二甲苯、二氯乙烷、氯仿、乙酸、乙酐、乙酸乙酯、已烷、四氢呋喃、三乙胺、吡啶、DMF 等溶剂。DMAP的分子结构,如图1-1。http://ng1.17img.cn/bbsfiles/images/2015/09/201509272125_568160_2423358_3.jpg1.2实验试剂规格及来源实验试剂规格及来源,均符合分析化学实验的要求,可以保证实验的进行。表1-2 实验所用试剂 试剂名称 生产厂家分子量规格和品级质量分数% DMAP 北京华威锐科化工有限公司122.17分析纯99.00 乙醇 北京化工厂46.07分析纯99.70 正丙醇 天津市福晨化学试剂厂60.10分析纯99.80 异丙醇 北京化工厂60.10分析纯99.70 正丁醇 北京化工厂74.12分析纯99.00 异丁醇 天津市福晨化学试剂厂74.12分析纯99.00 乙酸甲酯 天津市光复精细化工研究所74.08分析纯98.00 乙酸乙酯 北京化工厂88.11分析纯99.50 乙酸丙酯 天津市光复精细化工研究所102.13分析纯98.00 乙酸异丙酯 天津市光复精细化工研究所102.13分析纯99.00 乙酸丁酯 北京化工厂116.16分析纯99.00 乙酸异丁酯 天津市光复精细化工研究所116.16分析纯98.501.3实验装置表1-3 实验的仪器设备 设备名称 型号 生产厂家 精密温度计 制冷和加热循环槽 0-50℃ MPG-10C型 上海精密科学仪器厂 上海一恒科技仪器有限公司 电子分析天平 Sartorius CP124S型 德国Sartorius公司 磁力搅拌器 85-1A型 巩义市于华仪器有限责任公司 激光发射器 JDW-3型激光电源 北京大学物理系 夹套溶解釜 定制 北京化工大学仪器厂 本实验用到的仪器设备列于表1-3。 实验装置主要包括激光监视系统、夹套溶解釜、磁力搅拌器、电子分析天平、制冷和加热循环槽等。激光监视系统是发射激光、接受激光、记录仪等组成的,激光具有单色性好、相干性高、方向性强的特点,应用到测定溶解度,可减少因人为目测试样溶解情况带来的误差。夹套溶解釜是一个双层玻璃的瓶子,外层接通循环水,使内层试样升温或降温,还具有保温功能,内层装实验试样,内层上方有两个瓶口,大瓶口以插有温度计的塞子为塞子,大瓶口加入溶剂或溶质,小瓶口接冷凝管,冷却逸出液面到达瓶口处的试样,减少试样的挥发。磁力搅拌器充分搅拌瓶内试样,加快溶质的溶解。电子分析天平用来称量溶剂、溶质的质量。制冷和加热循环槽是一个超级恒温水浴系统,通过设定循环槽的温度调整溶解釜的温度,确保在加入溶质的实验过程中为恒定温度下。http://ng1.17img.cn/bbsfiles/images/2015/09/20150927212
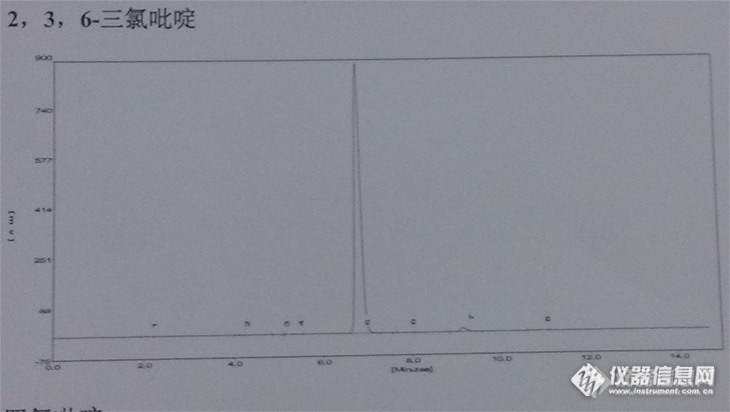
之前采用大连依利特液相色谱仪分析各类吡啶氯化物,色谱柱为Agilent TC-C18 250mm*4.6mm,采用0.1%的磷酸水溶液:CH3CN=30:70作为流动相,柱温为室温条件,流速为1.0ml/min,UV检测波长为235nm。谱图如下:吡啶http://ng1.17img.cn/bbsfiles/images/2013/03/201303222121_431872_2047060_3.jpg五氯吡啶http://ng1.17img.cn/bbsfiles/images/2013/03/201303222124_431873_2047060_3.jpg四氯吡啶http://ng1.17img.cn/bbsfiles/images/2013/03/201303222124_431874_2047060_3.jpg2,6-二氯吡啶http://ng1.17img.cn/bbsfiles/images/2013/03/201303222125_431875_2047060_3.jpg2,3,6-三氯吡啶http://ng1.17img.cn/bbsfiles/images/2013/03/201303222125_431876_2047060_3.jpg
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测二氯二甲吡啶酚用什么毛细柱?(KB-wax或KB-pesticides B可以么 )
我这里有几份分析资料,想和大家分享。内容含有分析条件及色谱图, 有需要的就下载吧!1. 分析食品中的丙酸2. 涂料中一般溶剂(二氯甲烷,乙苯_二甲苯)的分析3. 分析大蒜中的二烯丙基二硫(蒜素)
1.如何分析丙烯酸中的乙二醛,水中乙二醇? 乙二醛会导致丙烯酸聚合,应越少越好。乙二醇为抗冻剂,检查工业水中的乙二醇含量可以确认冷冻水管是否破裂,滲漏。1.曾以GC,LC,UV,GC-MASS进行尝试,均定性不出来。 GC上曾尝试DB-WAX,HP-5,FFAP等不同的管住进行分析。 LC的管住为SB-C8,加入不同浓度的标准品,峰形却几乎不变。波长扫描后,换波长也没用。 请教各位有什么高招进行分析。
请教老师:我想将乙醇、丙酮、乙酸甲酯、二异丙基胺、吡啶、二氯甲烷分开,您上次告知了柱温及方法,可是进样口温度、检测器温度、流速及分流比该怎么设定呢?多谢!
问:请问乙酰氯的GC分析方法极其标准?我们是是化工企业,生产高纯乙酰氯,分析量很大,我们目前用GC-FID毛细管柱子,没有标准品,含量大约99。5%,请问这样分析的缺陷,用HPLC行不行?假如有其他微量杂质,是否还要用质谱仪?
各位筒子们,液质分析4氨基吡啶,竟然木有检测到,N,N-二甲基吡啶倒是出峰了,这两个的结构比较相似,应该液质分析都没有问题,不知道原因出在哪里?
化验分析硅石标准品,怎样测铝的含量,标准品如何处理?







 我要推广仪器
我要推广仪器
 下载APP
下载APP