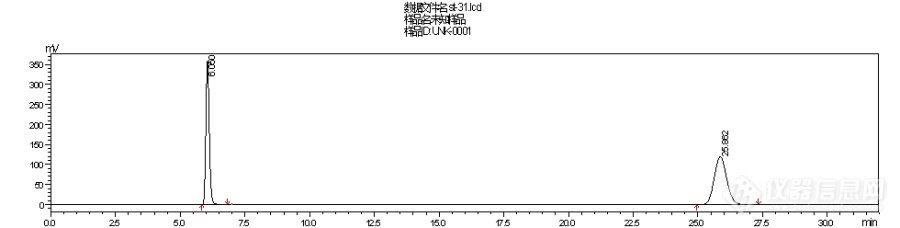
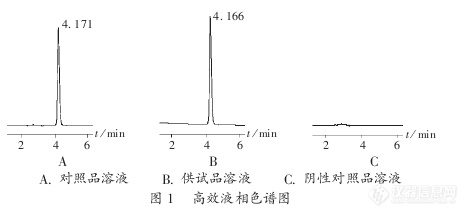
实验过程中参照标准进行,往往不能按照标准重复出来,根据药典标准要求对茜草含量进行分析,发现流动相的比例不太适应本实验室,对其进行方法摸索。1材料茜草药材(送检样);甲醇(分析纯),浓盐酸,三乙胺,磷酸;[color=#333333]大叶茜草素,羟基茜草素[/color][color=#333333]对照品(均购自中检院)[/color]。[color=#333333]2 [/color][color=#333333]仪器与设备[/color][color=#333333]岛津液相[/color][color=#333333]LC-20AT[/color][color=#333333](带紫外检测器及自动进样器);色谱柱安捷伦[/color][color=#333333]Zorbax SB C18(250mm*4.6μm*5μm)[/color][color=#333333];超声波清洗仪([/color]昆山市超声仪器有限公司[color=#333333]);[/color]DZKW-S-6型电热恒温水浴锅(北京市永光明医疗器械仪器有限公司)。3 实验过程(按照2015年版中国药典一部 茜草项下含量测定操作) 取本品粉末(过二号筛)约0. 5g,精密称定,置具塞锥形瓶中,精密加入甲醇100ml,密塞,称定重量,放置过夜,超声处理(功率250W,频率40kHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液50ml,蒸干,残渣加甲醇-25%盐酸(4 : 1)混合溶液20ml溶解,置水浴中加热水解3 0分钟,立即冷却,加入三乙胺3ml,混匀,转移至25ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。以甲醇-乙腈-0.2%磷酸溶液(25 : 50 :25)为流动相;检测波长为250nm。4 流动相条件[color=#333333](1)[/color]按照药典流动相条件[color=#333333]甲醇:乙腈:0.2%磷酸溶液(25:50:25),发现样品10min之前杂质较多,羟基茜草素受杂质干扰严重,调有机相比例但是依然不能完全分开,我们更换色谱柱,效果不佳。[/color][align=center][color=#333333][img=,690,174]https://ng1.17img.cn/bbsfiles/images/2019/07/201907181039432269_1092_3920249_3.png!w690x174.jpg[/img][/color][/align][align=center][color=#333333]药典标准流动相比例等度条件下的标准品色谱图[/color][/align][align=center][color=#333333][img=,690,174]https://ng1.17img.cn/bbsfiles/images/2019/07/201907181041183621_4282_3920249_3.png!w690x174.jpg[/img][/color][/align][align=center][color=#333333][color=#333333]药典标准流动相比例等度条件下的样品色谱图[/color][/color][/align][color=#333333](2)[/color][color=#333333]配成单相混合溶液也是羟基茜草素分离度不好。[/color][color=#333333](3)[/color][color=#333333]本实验室液相二元泵的多一些,我们为了减少流动相配比误差,将流动相条件改为两相走梯度洗脱,发现茜草中两种有效成分可以得到很好的分离,并且不受其他物质的干扰。乙腈与0.2%磷酸水梯度0min-40min [color=#333333]乙腈[/color]40%升至80%,40min-50min 80%乙腈。[/color][align=center][color=#333333][img=,690,174]https://ng1.17img.cn/bbsfiles/images/2019/07/201907181051229011_3007_3920249_3.png!w690x174.jpg[/img][/color][/align][align=center][color=#333333]乙腈和0.2%磷酸水梯度洗脱标样色谱图[/color][/align][align=center][color=#333333][img=,690,174]https://ng1.17img.cn/bbsfiles/images/2019/07/201907181051448461_9667_3920249_3.png!w690x174.jpg[/img][/color][/align][color=#333333][/color][align=center]乙腈和0.2%磷酸水梯度洗脱样品色谱图[/align][align=left] 检测中药的过程中发现大部分药材基质比较复杂,完全按照药典的比例不一定能有效分离,做研究的过程中,会对条件进行优化,以保证数据的准确性与可靠性![/align]
[size=16px][font=Arial, &][color=#333333]目的[/color][/font][font=Arial, &][color=#333333] 考察茜草炭炮制过程中饮片色度值与超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(UPLC)特征图谱的相关性。 [/color][/font][font=Arial, &][color=#333333]方法[/color][/font][font=Arial, &][color=#333333] 采用UPLC建立茜草及茜草炭的特征图谱,并采用分光测色仪测定其色度值:明暗度值(L*)、红绿色值(a*)、黄蓝色值(b*),对茜草炭炮制过程中饮片色度值与特征图谱进行相关性分析。 [/color][/font][font=Arial, &][color=#333333]结果[/color][/font][font=Arial, &][color=#333333] 随着炮制时间的延长,茜草炭较茜草饮片特征图谱的相似度逐渐降低 异茜草素、6-羟基甲基异茜草素单位峰面积先增大后减小,其余共有峰单位峰面积均呈减小趋势 L*、a*、b*值整体上逐渐减小,而△E*逐渐增大。L*、a*、b*值分别与羟基茜草素、6-羟基甲基异茜草素、茜草素呈极显著正相关。主成分分析共提取2个主成分,累积方差贡献率为88.292%。聚类分析结果显示,炮制0~6 min的样品聚为一类,炮制8~34 min的样品聚为二类。正交偏最小二乘法判别分析结果显示,6-羟基甲基异茜草素、异茜草素、a*、b*是茜草炭饮片炮制过程中发生质量变化的主要指标。 [/color][/font][font=Arial, &][color=#333333]结论[/color][/font][font=Arial, &][color=#333333] 不同炮制时间茜草炭饮片的色度值与UPLC特征图谱密切相关,可为茜草炭饮片炮制的在线监控和质量评价提供较全面的判断依据。[/color][/font][/size]
请问茜草含量方法中药典规定下的水浴水解具体怎么操作?是要回流吗?[img]https://ng1.17img.cn/bbsfiles/images/2022/08/202208281116087303_9982_5361118_3.png[/img]
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font][/b][font=宋体][/font][b][font=宋体][/font][/b] [font=宋体] [/font] [font=宋体][/font] [font=宋体](1)本品根横切面:木栓细胞6~12列,含棕色物。栓内层薄壁细胞有的含红棕色颗粒。韧皮部细胞较小。形成层不甚明显。木质部占根的主要部分,全部木化,射线不明显。薄壁细胞含草酸钙针晶束。[/font] [font=宋体](2)取本品粉末0.2g,加乙醚5ml,振摇数分钟,滤过,滤液加氢氧化钠试液1ml,振摇,静置使分层,水层显红色;醚层无色,置紫外光灯(365nm)下观察,显天蓝色荧光。[/font] [font=宋体](3)取本品粉末0.5g,加甲醇10ml,超声处理30分钟,滤过,滤液浓缩至1ml,作为供试品溶液。另取茜草对照药材0.5g,同法制成对照药材溶液。再取大叶茜草素对照品,加甲醇制成每1ml含2.5mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各5[/font][font=&]μ[/font][font=宋体]l[/font][font=宋体],分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-丙酮(4:1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。[/font] [font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][b][font=宋体][/font][/b] [font=宋体][/font] [b][font=宋体][/font] [font=宋体][/font] [font=宋体]水 分 [/font][/b][font=宋体] [/font][font=宋体]不得过12.0%(通则0832第二法)。[/font] [b][font=宋体]总灰分 [/font][/b][font=宋体] [/font][font=宋体]不得过15.0%(通则2302)。[/font] [b][font=宋体]酸不溶性灰分[/font][/b][font=宋体] [/font][font=宋体]不得过5.0%(通则2302)。[/font] [b][font=宋体]【浸出物】[/font][/b][font=宋体] 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用乙醇作溶剂,不得少于9.0%。[/font] [b][font=宋体]【含量测定】 [/font][/b][font=宋体]照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以十八烷基硅烷键合硅胶为填充剂;以甲醇-乙腈-0.2%磷酸溶液(25:50:25)为流动相;检测波长为250nm。理论板数按大叶茜草素、羟基茜草素峰计算均应不低于4000。[/font] [b][font=宋体]对照品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取大叶茜草素对照品、羟基茜草素对照品适量,精密称定,加甲醇分别制成每1ml含大叶茜草素0.1mg、含羟基茜草素40[/font][font=&]μ[/font][font=宋体]g[/font][font=宋体]的溶液,即得。[/font] [b][font=宋体]供试品溶液的制备 [/font][/b][font=宋体]取本品粉末(过二号筛)约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇100ml,密塞,称定重量,放置过夜,超声处理(功率250W,频率40kHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液50ml,蒸干,残渣加甲醇-25%盐酸(4:1)混合溶液20ml溶解,置水浴中加热水解30分钟,立即冷却,加入三乙胺3ml,混匀,转移至25ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。[/font] [b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]分别精密吸取对照品溶液10[/font][font=&]μ[/font][font=宋体]l[/font][font=宋体]与供试品溶液20[/font][font=&]μ[/font][font=宋体]l[/font][font=宋体],注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,即得。[/font] [font=宋体]本品按干燥品计算,含大叶茜草素([/font][font=&]C[sub]17[/sub]H[sub]15[/sub]O[sub]4[/sub][/font][font=宋体])不得少于0.40%,羟基茜草素([/font][font=&]C[sub]14[/sub]H[sub]8[/sub]O[sub]5[/sub][/font][font=宋体])不得少于0.10%。[/font] [font=宋体] [/font]
由于日本现增加对食品中呋喃硫威和禾草丹检测,那位大侠有方法共享一下,谢谢![em24]
请问做呋喃丹、草甘膦需要的柱后反应器是什么啊,做这两个项目要怎么做?
做硝基呋喃类化合物检测用的标准品大家都是用的什么呀?有标准中说的是用对照品,有标准中用的是代谢物,不知道这其中有没有什么区别呀?大家都根据哪个标准做的呢?
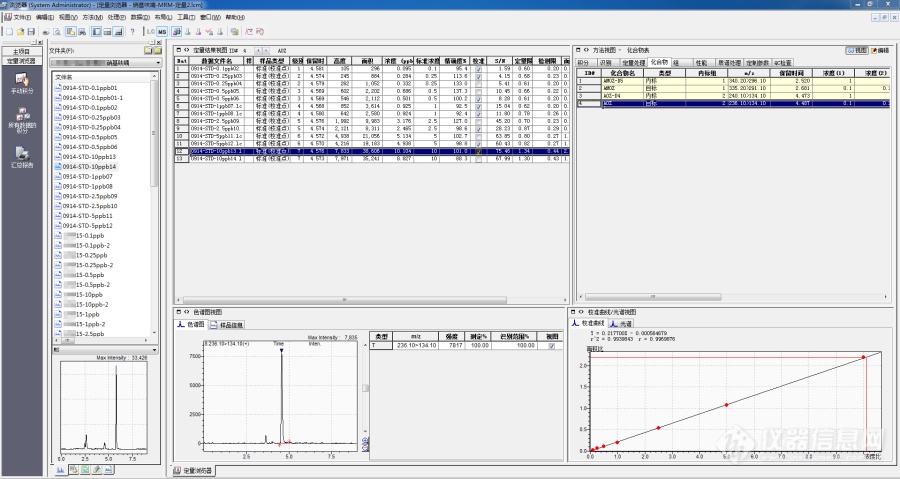
[b][b]液相色谱-串联质谱法测定草鱼中硝基呋喃类代谢物AOZ残留量[/b]摘要[/b]: 本文根据农业部783号公告-1-2006水产品中硝基呋喃类代谢物残留量的测定 液相色谱-串联质谱法来检测草鱼中硝基呋喃类代谢物AOZ的残留。样品经水解、衍生化和净化后,采用多反应监测模式测定。通过化合物的保留时间、定性离子和定量离子的筛查与确证,实现对草鱼中硝基呋喃类代谢物AOZ的定性与定量。结果表明,其线性系数大于0.99,在2.5mg/kg和4.5mg/kg添加水平下,AOZ回收率在95%-100%之间,相对标准偏差(RSD)小于15%。样品经水解、衍生化和净化后,以流动相A:0.002mol/L的醋酸铵溶液,B:甲醇进行梯度洗脱,C18(100mm×2.1mm,i.d,5um)色谱柱分离,采用正离子多反应监测( MRM) 模式检测,基质匹配标准溶液定量。线性范围内相关系数均大于[color=#ff0000] [/color]0. 99。加标回收率在95.00%-100% 之间,相对标准偏差在6.19%- 8.43% 之间,结果表明,该方法简便、快速、灵敏度高、重现性好,可测定草鱼中硝基呋喃类代谢物AOZ的残留。[b]关键词[/b]: 高效液相色谱 - 串联质谱 硝基呋喃代谢物AOZ 草鱼;[b]Abstract[/b]:In this paper, AOZ residue of nitrofuran metabolites in grass carp was detected by liquid chromatography-tandem mass spectrometry according to the determination of nitrofuran metabolites residues in aquatic products in the ministry of agriculture no.783 bulletin 1-2006. After hydrolysis, derivatization and purification, the samples were determined by multi-reaction monitoring. AOZ, a nitrofuran metabolite in grass carp, was qualitatively and quantitatively determined by retention time, qualitative and quantitative ion screening. The results showed that the linear coefficient was greater than 0.99, the recovery of AOZ was between 95% and 100% and the relative standard deviation (RSD) was less than 15% at the addition levels of 2.5mg/kg and 4.5mg/kg.After the samples were hydrolyzed, derivated and purified, the samples were separated by gradient elution in mobile phase A:0.002mol/L ammonium acetate solution,B: methanol, and separated by C18(100mm×2.1mm, i.d,5um) chromatographic column. Positive ion multireaction monitoring (MRM) mode was adopted for detection, and the matrix matched standard solution quantification. The correlation coefficients in the linear range are all greater than 0.99. The standard recovery was between 95.00% and 100%, and the relative standard deviation was between 6.19% and 8.43%.[b]Key words[/b]:ultra performance liquid chromatography-tandem mass spectrometry;nitrofuran metabolites-AOZ Grass carp.[b]1 实验部分[/b]1.1 仪器与试剂液相色谱—质谱联用仪( 岛津[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]-8030) 超纯水系统(WP-UP-YJ-20, 沃特浦) 分析天平(Quintix 2102-1CN,赛多利斯公司);旋涡混合器(VORTEX 3,IKA公司);恒温水浴振荡器(江苏安普SHA-C);离心机(湖南湘仪H-2050R);甲醇(色谱纯,默克股份两合公司) 醋酸铵( 分析纯,天津市致远化学试剂有限公司);2-硝基苯甲醛(色谱纯,天津市百世化工有限公司);二甲基亚砜(分析纯,天津市百世化工有限公司);磷酸氢二钾(分析纯,天津市百世化工有限公司);乙酸乙酯(分析纯,天津市泰兴试剂厂);农药标准物质: 均来自农业部环境保护科研监测所(天津);实验用水为经沃特浦超纯水系统处理后的超纯水。1.2 [b]样品处理[/b]1.2.1[b] 样品水解与衍生化[/b] 准确称取样品2.0g(精确至 0.01 g) 到50 mL 离心管中,加入0.05mL的100ng/mL混合内标(AOZ-D[sub]4[/sub]),旋涡混合50s,再加入5mL盐酸溶液(0.2mol/L)和0.15mL的2-硝基苯甲醛溶液(0.05mol/L),涡旋振荡50s后,置于恒温水浴振荡器中37℃避光震荡16h。1.2.2 [b]样品的提取净化[/b]取出离心管冷却至室温,加入3-5mL磷酸氢二钾溶液(1.0mol/L),调节pH至7.0-7.5,加入4mL乙酸乙酯,涡旋振荡50s,4000r/min离心5min,取上层清液转移至10mL玻璃离心管中;再加入4mL乙酸乙酯重复上述操作,合并上清液于40℃下氮气吹干。加入1.0mL甲醇溶液(甲醇:水=5:95(V:V))涡旋振荡溶解残留物,过0.45um滤膜,待测。1.2.3 [b]溶液配制 [/b](1)[b]标准储备液。[/b] 分别取上述标准品(100.0 ug/mL 溶液),用甲醇稀释成 10 ng/mL 和100 ng/mL的标准储备液。(2)[b]标准工作溶液。[/b] 分别吸取上述标准品10ng/mL的标准储备液0.010mL、0.025mL、0.050mL、0.10mL和100ng/mL的标准储备液0.025mL、0.05mL、0.10mL于7个50mL离心管中,不加样品,按照1.2.1和1.2.2步骤操作,按照1.3测定。1.3 [b]色谱 - 质谱条件[/b]1.3.1 [b]色谱条件 [/b] 色谱柱:C18柱,(100mm × 2.1mm,5μm ) 柱温为40℃;进样量为20μL。流动相:A.0.002mol/L的醋酸铵溶液,B:甲醇 色谱洗脱条件见表 1。[align=center]表 1 高效液相色谱梯度洗脱条件[/align][table][tr][td][align=center]时间,min[/align][/td][td][align=center]A,%[/align][/td][td][align=center]B,%[/align][/td][td][align=center]流速,mL. [/align][/td][/tr][tr][td][align=center]3.5[/align][/td][td][align=center]15[/align][/td][td][align=center]85[/align][/td][td][align=center]0.25[/align][/td][/tr][tr][td][align=center]5[/align][/td][td][align=center]15[/align][/td][td][align=center]85[/align][/td][td][align=center]0.25[/align][/td][/tr][tr][td][align=center]5.1[/align][/td][td][align=center]76[/align][/td][td][align=center]24[/align][/td][td][align=center]0.25[/align][/td][/tr][/table]1.3.2[b]质谱条件[/b]离子源: 电喷雾离子源ESI 扫描模式为正离子扫描 雾化气流量3L/min;干燥气流量15L/min;加热块温度250℃;DL温度250℃;CID气230kPa;IG真空度1.9×10[sup]-3[/sup]pa;PG真空度7.5×10[sup]1[/sup]pa;检测方式为多重反应监测。其他质谱参数见表2。 [align=center]表 2 反应监测的质谱采集参数[/align][table][tr][td][align=center]化合物[/align][/td][td][align=center]母离子m/z[/align][/td][td][align=center]子离子m/z[/align][/td][td][align=center]碰撞能量(v)[/align][/td][/tr][tr][td=1,2][align=center]AOZ[/align][/td][td][align=center]236[/align][/td][td][align=center]104[/align][/td][td][align=center]19[/align][/td][/tr][tr][td][align=center]236[/align][/td][td][align=center]134*[/align][/td][td][align=center]22[/align][/td][/tr][tr][td][align=center]AOZ-[/align][/td][td][align=center]240[/align][/td][td][align=center]134*[/align][/td][td][align=center]14[/align][/td][/tr][tr][td=4,1]注:*为定量碎片离子[/td][/tr][/table][b]1.4 添加回收实验[/b] 准确称取不含上述药物的草鱼样品2.0g,分别以2.5mg/kg和4.5mg/kg2个水平进行添加回收实验,重复5次,按照上述实验方法测定,计算添加回收率和相对标准偏差。[b]2 结果与讨论2.1线性回归方程[/b]AOZ的基质标准曲线为 y = 0.217700 x -0.000564679( r = 0.9969876,r[sup]2[/sup]=0.9939843) 如图3 [align=center]图3(标准曲线AOZ)[/align][img=图片3 标准曲线,690,367]https://ng1.17img.cn/bbsfiles/images/2019/10/201910100829478357_6345_3416090_3.png!w690x367.jpg[/img][b]2.2方法的准确度、精密度[/b]在空白草鱼中分别进行 2.5和4.5 mg / kg 2 个水平的加标回收试验,每个水平重复测定 5 次,计算加标回收率和相对标准偏差。由表 5 可知,在 2.5和4.5mg / kg 2个添加水平下,AOZ的平均回收率分别为 99.648% 和95.63% ,相对标准偏差分别为 8.43% 和6.19%,方法显示出良好的准确度和精密度,可以满足试剂样品中农药残留的检测要求。[align=center]表 5 AOZ在草鱼中的回收率和相对标准偏差[/align][table][tr][td][align=center]农药[/align][/td][td][align=center]添加浓度( mg / kg)[/align][/td][td][align=center]平均回收率( %)[/align][/td][td][align=center]重复 1[/align][/td][td][align=center]重复 2[/align][/td][td][align=center]重复 3[/align][/td][td][align=center]重复 4[/align][/td][td][align=center]重复 5[/align][/td][td][align=center]平均值相对标准偏差( % )[/align][/td][td][align=center]标准曲线[/align][/td][td][align=center]相关系数r2[/align][/td][/tr][tr][td]AOZ[/td][td]2.5[/td][td]99.648[/td][td]2.517[/td][td]2.261[/td][td]2.663[/td][td]2.723[/td][td]2.292[/td][td]8.43[/td][td][align=center]Y=0.217700X-0.000564679[/align][/td][td]0.9939843[/td][/tr][tr][td]AOZ[/td][td]4.5[/td][td]95.63[/td][td]4.223[/td][td]4.768[/td][td]4.043[/td][td]3.870[/td][td]4.613[/td][td]6.19[/td][td][align=center]Y=0.217700X-0.000564679[/align][/td][td]0.9939843[/td][/tr][/table][b]2.3实际样品分析[/b] 用本方法对市场销售的2份草鱼进行硝基呋喃类代谢物AOZ检测,均未检出,见图6。 [align=center]图6[/align][img=图片6,690,366]https://ng1.17img.cn/bbsfiles/images/2019/10/201910100830145934_7979_3416090_3.png!w690x366.jpg[/img][b]3、结论[/b]本研究通过对样品前处理方法、质谱条件和色谱条件的优化,建立了液相色谱-串联质谱法测定草鱼中AOZ残留量的分析方法,该方法前处理简单、快速、回收率高,方法的灵敏度、准确度和精密度等均满足农药残留分析的要求,适用于大量样品的快速检测。[b]参考文献[/b]高洁,朱莉萍等.超高效液相色谱-串联质谱法测定多脂肪类动物源性食品中硝基呋喃代谢物. 食品安全质量检测学报2018年第9卷第6期,2018.[b] [/b]
[color=#444444]我所使用的是Agilent LC/MS QTOF,想对样品中的一些呋喃香豆素类物质(没有购买到标准品)进行定性,分子量在210-800之间。我先做了一级质谱。能不能达到这样的目的:LC/MS上有跑出来的液相色谱的图,能不能根据一级质谱中的EIC图来确定或推断液相色谱的图上的某个峰是什么物质?[/color][color=#444444] 在自动提取EIC的图上有个保留时间,这个保留时间和液相色谱的保留时间有什么关系啊?看一个分子量如727.3110的EIC图,怎么会有好几个峰啊?有的同个分子量的有好几个保留时间,不知道是为什么?请大家给指点下哈!谢谢![/color]
请教下,我们做生食水产中硝基呋喃,是按照GB/T 21311-2007 动物源性食品中硝基呋喃类药物代谢物残留量检测方法 高效液相色谱/串联质谱法 ,我昨天在配置好的对照品溶液中,加了大概10倍量的邻硝基苯甲醛,37度过夜衍生,结果今天连母离子都找不到,想请教下各位是怎么做的。
大家好: 我在做食品中呋喃妥因代谢物时,不知道什么原因,所有的样品都检出,标准品也不成线性,回收率很高有时能达到好几百,请问呋喃妥因代谢物检测过程中有什么因素可以影响?谢谢指点。(我用的是Thermo的TSQ Quantum [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url])
10号走呋喃代谢物样品的时候,发现了一个很奇怪的现象,标准品保留时间正常,走空白和样品时AMOZ、SEM、AHD、AOZ四项内标保留时间全部偏离0.2min左右,最后一针标准品也没能幸免。可以肯定不是流动相的问题,柱子平衡时间也足够长。为了确认,又重新进了一遍,仍然重复上述情况。最后,把空白舍去,按照标准品、样品、标准品顺序运行,结果正常,空白的影响力这么大?一针空白可以导致后面序列全部异常(总共6针)?我真是第一次碰到这种奇怪的现象,问题的原因还没有找到,等看一下今天的样品结果再做判断吧。仔细看了一下,发现空白没有内标,测了一下空白的PH,大约在5-7,标准和样品都在3左右,难道真的是弄错了空白?查看了当天的样品,同时检测的还有克伦特罗、孔雀石绿、氯霉素和盐霉素,前三个都有内标监控,所以肯定没错,最有可能的是和盐霉素空白混了,想重新检测比较一下,结果进样小瓶已经处理掉了,哎,只能等有时间再验证了。17和18号进行了相关验证,发现使用盐霉素空白并不能导致呋喃代谢物保留时间漂移,看来当时的呋喃空白应该是预处理操作的时候出现了错误,因无法重现,可能要成为不解之谜了。。。。。。以下是当天保留时间偏离的三个样品和标准的TIC图谱,其中第一、三、五、七通道分别是AMOZ、SEM、AHD、AOZ的内标,每一通道前三个是样品,最后一个是标准品。大家看一下有没有遇到这样偏离的情况,讨论一下。http://ng1.17img.cn/bbsfiles/images/2010/11/201011202027_260858_1855403_3.gif
广州市食安办对散装熟食抽检时发现,好又多超市的1批次散装烤鸡检出违禁药物呋喃唑酮的代谢物AOZ。呋喃唑酮是硝基呋喃类药物的一种,不时在水产品中被检出。以往抽检结果的发现▲ 2014年8月,深圳市食药局开展“清水”专项执法监督抽查行动对水产品质量安全进行抽检。 ● 抽查了121批次水产品,其中2批次淡水鱼检出硝基呋喃代谢物(详情可回看我们微信平台9月4日的信息)。▲ 2013年9月,深圳市食品安全监管局开展水产品批发市场专项执法抽检。 ● 抽检结果显示,罗湖区一水产批发行中黄骨鱼内检出硝基呋喃代谢物。▲2013年9月,青岛市食安办公布流通领域水产品抽检结果。 ● 抽查了140批次水产品,其中2批次检出呋喃唑酮代谢物(AOZ)超标。硝基呋喃类药物 呋喃唑酮与呋喃西林、呋喃妥因、呋喃它酮四种药物统称为硝基呋喃类药物,是广谱抗菌药。 硝基呋喃类药物不稳定,动物摄入后容易在体内生成对应的四种代谢产物,从而残留在动物源性食品中,并通过食物链传递给人类。硝基呋喃类药物及其代谢物http://mmbiz.qpic.cn/mmbiz/GnnTib8oQtKAyZbYiaI2srzGTWCAEXfO5obP7RNtJqDwYYMFiahdbXYkQUBDU1ll5WD0ZrHwy3787WbB2CZia1RCzw/0主要国家和地区的规定 上世纪90年代,硝基呋喃类药物被发现对人体有一定毒副作用,目前已被多个国家和地区列为禁止在食品和动物中使用的兽药。1美国 FDA“联邦法规21章”(21CFR530.41)规定食源性动物禁止使用呋喃唑酮和呋喃妥因。2欧盟 (EC) No 470/2009《动物源食品中药理活性物质的残留限量》规定动物源性食品中不得检出硝基呋喃类药物。3韩国 2002年版《食品公典》规定猪肉中不得检出呋喃唑酮。 4中国 2002年12月24日发布的中华人民共和国农业部公告第235号及2005年10月28日发布的中华人民共和国农业部公告第560号规定,硝基呋喃类药物为在饲养过程中禁止使用的药物,在动物性食品中不得检出。2010年3月22日卫生部发布的《食品中可能违法添加的非食用物质名单(第四批)》中,明确将硝基呋喃类药物呋喃唑酮、呋喃它酮、呋喃西林、呋喃妥因列为非食用物质。http://mmbiz.qpic.cn/mmbiz/MVPvEL7Qg0EPD5jh0tV8WnQUh5DoR7UqXe88UP9HuYEcO8vV2H4JYaRAAF8m0dg1916mgEu3ia5MggE6TGEkDog/640 给企业的提示♥ 提高警惕,加强原料的质量监控,对动物源性食品,可将违禁兽药纳入重点监控因素。♥ 可要求供货商提供权威机构出具近期的、覆盖违禁兽药的产品合格证明材料。♥ 加强原料的自我检验工作,如企业自身无检测能力,可委托有资质的检测机构进行检验。
动物源性食品中硝基呋喃类药物检测的固相萃取方法一、实验目的本实验利用固相萃取法作为样品的前处理方法,LC-MS/MS法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂用量。二、实验目标物呋喃唑酮(CAS:67-45-8),呋喃它酮(CAS:139-91-3),呋喃西林(CAS:59-87-0),呋喃妥因(CAS:67-20-9)。三、应用范围本方法适用于动物源性食品中硝基呋喃类药物的LC-MS/MS检测及确证。四、参考文献 推荐性国家标准《GB/T 21311-2007 动物源性食品中硝基呋喃类药物代谢物残留量检测方法 高效液相色谱-串联质谱法》。五、实验材料 C8/SAX固相萃取柱200mg/6mL。六、实验方法1、样品前处理 将样品组织搅碎、均质。精确称取约1g(精确到0.01g)样品于15mL带螺盖的离心管中,加入1mL水和8mL甲醇,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液;加入8mL乙醇,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液;加入8mL乙酸乙酯,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液。2、水解和衍生化向质控和样品同待测样品中加入5mL 0.2mol/L的盐酸水溶液、100μL衍生化试剂、以及100μL内标工作溶液(20μg/L)和的混合标准溶液(10μg/L)。盖好盖子,充分振荡混匀,然后放入空气浴摇床,在37℃,200r/min条件下衍生化16h(过夜)。3、样品提取 加入0.3M的磷酸钠水溶液500μL。用试管滴加10mol/L的氢氧化钠溶液,涡旋混合均匀,用精密pH试纸调节pH值至7.0—7.2。9500r/min 离心10min,取上清液。4、SPE柱净化(1)活化:依次以5mL甲醇和5mL纯水预处理。(2)洗脱:上清液全部过柱,流速控制在约每秒1滴。依次以5mL水和5mL 50%甲醇/水溶液淋洗柱子。淋洗液完全通过小柱后,至少抽真空5min。以4mL 4%的甲醇氨洗脱,洗脱液用15mL试管收集。(3)浓缩定容:40℃氮气吹干。残渣以0.05%甲酸/甲醇溶液(9:1,v/v)溶解。溶解液以0.22μm的水相滤膜过滤,滤液可直接用于LC-MS/MS分析。5、LC-MS/MS条件 液相色谱-质谱/质谱仪 色谱柱:C18柱:150mm×2.1mm,2.0μm,或相当者 流动相:甲醇-5mM乙酸铵七、实验结果1、添加回收结果 向样品中加入不同水平的四环素类药物,回收率结果如下:(见表1)表1 动物组织中四环素类药物添加回收结果 样品名称 化合物名称 添加水平(ng/mL) 回收率(%) 猪肉 呋喃唑酮 50 80.75 100 82.58 呋喃它酮 50 89.74 100 90.88 呋喃西林 50 92.74 100 91.28 呋喃妥因 50 95.63 100 96.94 2、 空白样品添加农药残留物色谱图 http://ng1.17img.cn/bbsfiles/images/2015/08/201508141653_560805_3310_3.jpg
单位开展这个项目,但是目前没有呋喃代谢物的标准对照品,问了几家供应商都没有,不知道大家的都在什么地方买的,有没有电话可以联系
GBT 24800.5-2009 化妆品中呋喃妥因和呋喃唑酮的测定 高效液相色谱法
气中的四氢呋喃,我们用填充柱作的,在去年10月份作的时候保留时间是3.5分钟,后来就一直这样做的,也没什么问题。前不久,采的管道中的四氢呋喃,出峰时间漂移到1.5分钟,而3.5分钟没有峰,我们认为结果是未检出,客户提出疑问,说他们的污染源就是四氢呋喃,怎么会未检出呢?无奈重新进标液,奇怪的事情发生了,标液出峰时间也漂到1.5分钟,也就是说四氢呋喃现在出峰时间突然就变成1.5分钟了,而只有5分钟的运行时间,峰就漂移了2分钟,这以前从来没碰到过。 这个问题找不到原因,我个人分析是1)首先想到的是柱子脏了?进样口脏了?2)样品浓度太大了? 3)柱子坏了? 然而这些都没有找到支持的理由。求达人帮忙!
大家好,请问甲硝唑、氯霉素和呋喃唑酮都属于抗生素类兽药吗?中国食品产业网上有这样的分类:1.激素类 如已烯雌酚及其盐、酯和制剂,醋酸甲孕酮及制剂,甲基睾丸酮,丙酸睾酮,氯丙嗪,安定等;2.抗生素类 如氯霉素,呋喃唑酮,甲硝唑等;3.消毒和杀虫剂类 如五氯酚钠,孔雀石绿,硝酸亚汞等。 请问是正确的吗?谢谢各位了!
求呋喃能力验证过的大神支个招。实验室正在做呋喃的测量审核,因为前两次能力验证的实测值都大于120%,没过,一直在纠结,我们是不是衍生试剂2硝基苯甲醛加大4倍,还是PH调不对。按照农业部783号公告做的。昨天做的最后一步去脂肪加入定容液加正己烷的时候,只有考核样乳化了,其他质控样完好。白做了,总共7天时间。如果这次再不过真的要被骂了。求各位大神支个招。
新手,第一次做食品中硝基呋喃类药物的检测。请做过的老师们指点一下。用硝基呋喃类代谢物标准品优化质谱参数还需要衍生么,样品前处理衍生的目的是什么? 衍生的话,它们的母离子跟子离子质量数还跟国标上一致么?国标21311里面衍生,不同浓度的基质标曲加入的衍生剂的量相同。这样会不会影响衍生的效果?
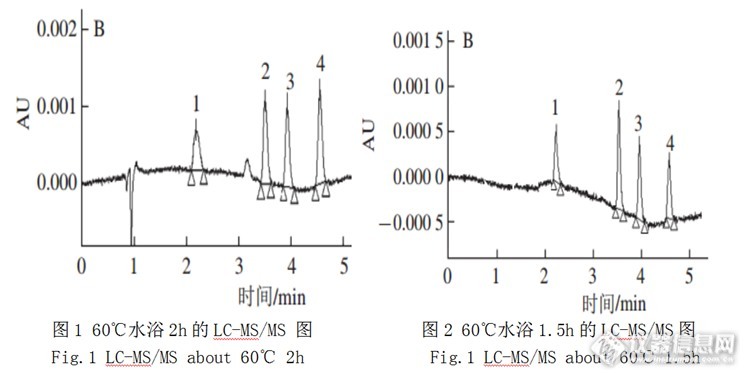
本人对肉粉这种基质复杂的样品进行处理,同时结合硝基呋喃代谢物提取时间长,提取率不高的问题进行优化,最终确定最佳的条件。[align=center][b]肉粉中硝基呋喃代谢物的高速动能前处理方法[/b][/align][align=center]王成梅[/align][align=center](山东中质华检测试检验有限公司,山东省 济宁市 邮编272000;)[/align][b]摘要:[/b]采用高速动能前处理技术,建立了快速、提取率高、回收率好的肉粉中硝基呋喃代谢物的液相色谱-串联质谱方法。处理后的样品添加同位素内标、酸水解、2-硝基苯甲醛衍生化、乙酸乙酯提取,正己烷除油,采用C18色谱柱进行分离,乙腈-0.002mol/L的乙酸铵为流动相进行梯度洗脱,多反应监测(MRM)模式检测。结果表明,采用高速动能处理过的样品在60℃经过2h衍生后,四种硝基呋喃代谢物在各自的线性范围内线性关系良好,相关系数均大于0.9990;方法的检出限在0.2ug/kg;通过三个低、中、高水平的加标,平均回收率(n=3)为91.7-105.2%,标准偏差小于3.10%。本方法采用一种新型的处理样品的方法,缩短了时间,提高了效率,增加了回收率,适合于基质复杂的肉粉中硝基呋喃代谢物的测定。关键词:高速动能;硝基呋喃代谢物; 肉粉; 液相色谱-质谱方法Quantitative Determination of Four Nitrofuran Metabolites in Meat Meal using high speed kinetic energy pretreatment technology[b]Abstract[/b]:Using high speed kinetic energy pretreatment technology, a method for the simultaneous determinatin of four nitrofuran metabolites in meat meal by liquid chromatography tandem mass spectrometry([url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)was eatalished.The method has a rapid ,high extraction and recovery rate.The homogenized sample was spiked with isotope inter standards,hydrolyzed withHCl,derivatized with nitrobenzaldehyde,extracted with ethyl acetate,and degreasing by hexane.The analysis was carried out on C18 column by gradient elution with acetonitrile-ammonium acetate as mobile phase,and detected by MS/MS system with positive electrospray ionization under multiple reaction monitoring(MRM)mode.The compounds was identified with retention time and ion ratio and quantified by the internal standard calibration curve isotope dilution technique.After 60℃ derivation,the results showed that the correlation coefficients(R20.9990)of four targets were obtained within their respective linear ranges over dynamic range of 0.2-5μg/L .The average recoveries were ranged from 91.7%-105.2% for the four targets at three spiked levels with the relative standard derivation (RSD,n=6)were under 3.10%.The method was precise and sensitivity,suitable for quantitative and qualitative analysis of four nitrofuran metabolites in meat meal.[b]Key words[/b]:High-speed kinetic energy nitrofuran metabolites meat meal [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS 硝基呋喃类药物是一种光谱抗生素,对大多数革兰氏阳性菌和革兰氏阴性菌一及真菌等都有杀菌作用。硝基呋喃类代谢物广泛应用于畜禽和水产养殖,以治疗有大肠杆菌或沙门氏菌引起的肠炎、溃疡病等。硝基呋喃类药物在动物体内半衰期短,但是其代谢物可以和蛋白质紧密结合,残留时间长,甚至于经过高温、微波、烘烤等处理也无法降解[sup][/sup]。研究表明,硝基呋喃类及其代谢物对人体具有致癌、致畸胎副作用,欧盟已于1995年禁止在食用动物中添加硝基呋喃类药物[sup][/sup],中国卫生部在2010年就将硝基呋喃类药物硝基呋喃唑酮、呋喃它酮、呋喃妥因、呋喃西林列入违禁添加物质黑名单。对于硝基呋喃类药物的检测不能反映其在动物性体内真实的情况。目前对于硝基呋喃类药物的检测主要是对其代谢物的检测,主要方法有免疫分析法[sup][/sup]、液相色谱法[sup][/sup]、液相色谱-质谱法[sup][/sup]。液相色谱-质谱法灵敏度和选择性都比较高,对于基质复杂样品的检测具有明显的优势,是目前最常用的检测方法。 细胞的破碎与分离是代谢物的提取和分析中的重要步骤。高速动能处理技术是近一两年发展起来的一种新型的高科技的前处理方法。它通过一种极速的均质技术将鱼粉进行细胞破碎与分离,便于代谢物的提取。 鱼粉是由新鲜鱼类经过酶解或者蒸煮、胶磨、均质、喷雾干燥、过筛等一系列的程序生产的食品配料。其最大的特点是可以溶于水,既保留了天然鱼肉的风味,又富含营养。经过研究表明,鱼粉中粗蛋白的含量在50%以上,粗脂肪10%左右,氨基酸含量在40%-78%左右,其中人体必需氨基酸甘氨酸、精氨酸和脯氨酸含量较高。由于鱼粉在加工的过程中添加盐、防腐剂、增香剂等,导致样品基质复杂,在进行提取检测的过程中会出现分层不明显,油脂含量高等现象,难以保证检测过程中的准确性和灵敏性。目前关于鱼粉中硝基呋喃类代谢物的前处理几乎不涉及对样品的处理,因此建立一种新型的鱼粉高速动能处理技术对于硝基呋喃类代谢物的检测具有重要的研究价值和意义。本文通过高速动能前处理技术,同位素内标法,高温水解衍生快速处理。乙酸乙酯提取,正己烷除油,多重反应检测处理,从而建立一种样品前处理简单、提取速度快、净化效果好、选择性和灵敏度高的鱼粉中硝基呋喃代谢物的新型检测方法。[b]1实验部分[/b]1.1 主要仪器和试剂 Agilent1260-6460液相色谱-质谱联用仪(美国安捷伦公司);高速动能仪(美国);Sigma3K15离心机(德国);IKA RV10旋转蒸发仪(德国);恒温水浴锅;涡旋混合器XW-80A(中国);电子天平Secura224-1cn(瑞士);pH计;Milli-Q去离子水发生器(美国Millipore公司)。 硝基呋喃类药物的标准品;3-氨基-2-唑烷基酮(AOZ)、5-甲基吗啉-3-氨基-2-唑烷基酮(AMOZ)、1-氨基-2-内酰脲(AHD)、氨基脲(SEM)以及各自的同位素内标(纯度大于99%,均购至德国Dr.Ehrenstorfer GmbH);2-硝基苯甲醛;甲醇、乙腈;甲酸铵、甲酸(HPLC级,);乙酸乙酯、正己烷、四氯化碳、氢氧化钠、盐酸、磷酸氢二钾、磷酸二氢钾均为分析纯;超纯水(18.2ΜΩ)。1.2 标准溶液的配制 四种硝基呋喃代谢物标准贮备液:准确称取适量的硝基呋喃代谢物标准物质用乙腈配成1.0mg/ml的标准储配液,-18℃冷冻避光保存,有效期为三个月。四种硝基呋喃代谢物混合标准工作液(1.0μg/ml):取适量的硝基呋喃代谢物的标准贮备液用乙腈稀释成1.0μg/ml的混合工作使用液,-18℃冷藏避光保存,有效期为一个月,使用前回到室温左右。 氘代内标混合液储备液(0.1mg/mL):分别准确氘代内标物用乙腈溶解,配制成0.1mg/mL的标准储备液,-18℃冷冻避光保存,有效期为6个月。 氘代内标混合工作使用液(1mg/L):准确吸取一定量的氘代内标混合工作储备液用乙腈稀释成浓度为1mg/L的混合工作使用液,冷藏避光保存,有效期为1个月。 2-硝基苯甲醛衍生溶液:准确称取0.15g 2-硝基苯甲醛,用甲醇定容至10mL,现用现配。1.3 样品的前处理1.3.1 样品的水解和衍生 取一定的样品经过高速动能仪进行60s高速均质破碎。称取1.0g经过高速动能破碎的样品于50ml的离心管中,加入10ml的0.125mol/L的盐酸溶液,涡旋混匀1min,加入100ul2-硝基苯甲醛衍生试剂60℃恒温水浴衍生2h。1.3.2 提取 取出样品,冷却至室温。用2mol/L的氢氧化钠和1mol/L的磷酸氢二钾调节pH值至7.2左右。离心,收集上清液加入15ml的乙酸乙酯震荡30min,离心,转移上清液至另一离心管中,残渣继续用乙酸乙酯提取一次,合并清液。向上清液中加入15ml的正己烷震荡10min,离心,弃去正己烷层,重复上述操作一次。最后收集的清液于40℃条件下旋转蒸发至干,用1ml甲酸(0.2%)2.5ml的液态混合净化剂(正己烷:四氯化碳=1:1)转移至离心管中,超声,涡旋,离心,上清液过0.22μm滤膜过滤上机待测。1.3.3 UPLC色谱条件 Agilent超高效液相色谱柱:ZORBAX Eclipse Plus C[sub]18[/sub](1.8μm,2.1mm×50mm) 柱温:30℃;进样体积:2μL;流速:0.4ml/min;后运行:2min。梯度洗脱条件见下表1[align=center]表1 液相色谱洗脱条件[/align][align=center]Table 1 HPLCgradient elution program[/align][table][tr][td][align=center]时间(min)[/align][/td][td][align=center]A(0.1%甲酸水含乙酸铵)[/align][/td][td][align=center]B 乙腈[/align][/td][/tr][tr][td][align=center]0[/align][/td][td][align=center]90[/align][/td][td][align=center]10[/align][/td][/tr][tr][td][align=center]3.0[/align][/td][td][align=center]70[/align][/td][td][align=center]30[/align][/td][/tr][tr][td][align=center]5.1[/align][/td][td][align=center]0[/align][/td][td][align=center]100[/align][/td][/tr][tr][td][align=center]8.1[/align][/td][td][align=center]0[/align][/td][td][align=center]100[/align][/td][/tr][tr][td][align=center]8.2[/align][/td][td][align=center]90[/align][/td][td][align=center]10[/align][/td][/tr][/table]1.3.4 质谱条件 离子源:电喷雾离子源;监测方式:多重反应检测;毛细管电压:4000V;离子源温度:350℃;脱溶剂气压力:40psi;脱溶剂气流量:11 L/min。质谱条件见下表2。[align=center]表2 硝基呋喃代谢物MRM 分析参数[/align][align=center]Table 2 Analysis parameters of nitrofuran metabolites class MRM[/align][table][tr][td][align=center]project[/align][/td][td][align=center]parent ion[/align][/td][td][align=center]Daughter ion[/align][/td][td][align=center]energy/v[/align][/td][td][align=center]fragmentor/v[/align][align=center] [/align][/td][td][align=center]pattern[/align][/td][td][align=center]acceleration voltage/v[/align][/td][/tr][tr][td][align=center] AOZ[/align][/td][td][align=center]236.1[/align][/td][td][align=center]133.8[/align][/td][td][align=center]7[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AOZ[/align][/td][td][align=center]236.1[/align][/td][td][align=center]103.8[/align][/td][td][align=center]17[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]AOZ-D4[/align][/td][td][align=center]240[/align][/td][td][align=center]134[/align][/td][td][align=center]7[/align][/td][td][align=center]100[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AMOZ[/align][/td][td][align=center]335.1[/align][/td][td][align=center]291.2[/align][/td][td][align=center]6[/align][/td][td][align=center]110[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AMOZ[/align][/td][td][align=center]335.1[/align][/td][td][align=center]261.9[/align][/td][td][align=center]12[/align][/td][td][align=center]110[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]D5-AMOZ[/align][/td][td][align=center]340[/align][/td][td][align=center]296[/align][/td][td][align=center]8[/align][/td][td][align=center]110[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AHD[/align][/td][td][align=center]249[/align][/td][td][align=center]133.9[/align][/td][td][align=center]6[/align][/td][td][align=center]100[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AHD[/align][/td][td][align=center]249[/align][/td][td][align=center]104[/align][/td][td][align=center]18[/align][/td][td][align=center]100[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AHD-13C3[/align][/td][td][align=center]252[/align][/td][td][align=center]134[/align][/td][td][align=center]7[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]SEM[/align][/td][td][align=center]209[/align][/td][td][align=center]192[/align][/td][td][align=center]6[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] SEM[/align][/td][td][align=center]209[/align][/td][td][align=center]165.9[/align][/td][td][align=center]9[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]SEM-C3,15N2[/align][/td][td][align=center]212[/align][/td][td][align=center]168[/align][/td][td][align=center]4[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][/table][b]2结果与分析[/b] 2.1样品处理 鱼粉是一种基质复杂的粉状物质,大多数实验室是不经过样品处理的。硝基呋喃类在体内代谢迅速,代谢的部分化合物分子与细胞膜蛋白结合成结合态,也就是所谓的硝基呋喃代谢物。该代谢物比较稳定可以长期保持稳定状态,从而可以在人体内长期驻留。普通的处理很难将硝基呋喃类药物和蛋白质分离。本实验通过一种新型的高速动能仪是样品在极短的时间可以进行高速的均质使样品高速分散,造成组织和细胞破碎,代谢物的流出,有利于提取。对比经过高速动能处理后的样品的提取率和回收率,数据表明经过处理的样品很少出现乳化现象,提取率也高。本实验采用高速动能处理时间短、提取率高、很大程度避免了基质复杂提取过程中造成的乳化现象,非常适合于鱼粉中硝基呋喃代谢物类的检测。2.2 酸解、衍生化 四种硝基呋喃代谢物分子质量相对比较小,进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析时,在这个质量轴范围内易受低端分子离子的影响,同时这一段背景干扰比较大,对定性和定量分析造成一定程度的干扰。硝基呋喃代谢物在酸性条件下可以和蛋白质分离加入2-硝基苯甲醛衍生试剂后可以和其结合形成比较大的分子团避开小分子离子的干扰,获得较高的灵敏度和特异性。而衍生需要在一定的温度和时间条件下才可以,目前最常用的是37℃水浴振摇16h,耗时长,出现问题给检测工作造成很大的困扰。本实验对比不同的水浴温度和水浴时间,最终确定最佳条件,节省了检测时间 (见图 1)。[img=,690,349]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020909_01_2984502_3.png[/img]2.3线性和回收率 在对复杂基质进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析时常会出现基质效应,影响定量分析的准确性和精明度。本实验采用同位素内标法减少了外标法过程中存在的干扰,使定量变得更准确。分别对0.2μg/L、0.5μg/L、1.0μg/L、2.0μg/L、5μg/L的基质混合标准溶液进行测定,四种硝基呋喃代谢物的线性范围、线性方程、相关系数、检出限和定量限见下表3。结果表明:四种代谢物线性关系良好,检出限为0.3μg/L,相关系数均大于0.990。分别对低、中和高三个浓度进行加标回收,按照本方法进行提取、衍生和测定,平行测定3此,计算其回收率。采用统一添加水平的样品连续测定6次,计算其稳定性。结果见表4,四种硝基呋喃代谢物的平均回收率在91.7-105.2%之间,相对偏差小于3.10%,该方法适用性良好。[align=center]表3 鱼粉中四种目标物的线性范围、线性方程、相关系数、检出限和定量限[/align][align=center]Table Linear ranges,regression equations,LODs and LOQs for 4 analytes in samples[/align][table][tr][td]Analyte[/td][td]Linear ranges/(μg/L)[/td][td]regression equations[/td][td]R2[/td][td]LOD/(μg/Kg)[/td][td]LOQ/(μg/Kg)[/td][/tr][tr][td]AOZ[/td][td]0.2~5[/td][td]y=0.822736x-0.006286[/td][td]0.9997[/td][td]0.05[/td][td]0.20[/td][/tr][tr][td]AMOZ[/td][td]0.2~5[/td][td]y=2.515006x-0.022236[/td][td]0.9989[/td][td]0.05[/td][td]0.20[/td][/tr][tr][td]AHD[/td][td]0.2~5[/td][td]y=0.837220x+0.049223[/td][td]0.9997[/td][td]0.20[/td][td]0.50[/td][/tr][tr][td]SEM[/td][td]0.2~5[/td][td]y=2.210855x+0.033769[/td][td]0.9967[/td][td]0.20[/td][td]0.50[/td][/tr][/table][align=center]表4 样品的添加回收和精密度实验数据[/align][align=center]Table 4 Recoveries and repeatabilities for 4 analytes in sample[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Added/(μg/kg)[/align][/td][td][align=center]Recovery/%[/align][/td][td][align=center]RSD/(%,n=6)[/align][/td][/tr][tr][td][align=center]AOZ[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]91.7,94.3,97.2[/align][/td][td][align=center]2.34,2.56,2.92[/align][/td][/tr][tr][td][align=center]AMOZ[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]92.0,93.1,99.3[/align][/td][td][align=center]2.41,2.85,2.91[/align][/td][/tr][tr][td][align=center]AHD[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]94.6,98.7,105.2[/align][/td][td][align=center]2.69,2.19,3.10[/align][/td][/tr][tr][td][align=center]SEM[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]82.9,95.9,96.9[/align][/td][td][align=center]2.58,2.34,2.90[/align][/td][/tr][/table][b]3结论[/b] 本方法通过引用一种新型的处理样品的技术高速动能仪,使样品在极短的时间里达到高速分散,不仅可以提高提取率而且避免了后续提取过程中出现的乳化现象。本实验称取经过高速动能仪处理过的样品加入同位素内标,加酸使呋喃代谢物和蛋白质分开,加入衍生剂2-硝基苯甲醛在60℃水浴条件下震荡衍生2h,调pH值,正己烷除油,乙酸乙酯提取,液态净化剂净化,液相色谱-串联质谱分析,建立了检测鱼粉中四种硝基呋喃代谢物的高速动能处理方法。该方法采用一种新型的分散均质技术、优化水解衍生条件,具有耗时短、处理过程简单、提取率高、灵敏度好、检出限低等优点,能够满足国内外对硝基呋喃代谢物限量的要求。
大家都用的是什么标准的方法?我用的是: 氯霉素:NY5070--2002,无公害食品 水产品中渔药残留限量 附录A 己烯雌酚 SC/T3020-2004水产品中己烯雌酚残留量的测定 酶联免疫法 呋喃唑酮代谢物 DB32/T 1038-2007 水产品中呋喃唑酮代谢物残留量的测定有不同的吗?我是江苏的,呋喃唑酮代谢物测定时用的地标。
使用以下化学品,对身体的危害大么?(正己烷,环己烷,丙酮,四氢呋喃,异丙醇) 由于不是化学专业的,有以上化学品的MSDS,但足以把自己看糊涂了;请问以上化学品究竟有多毒?需要怎样的防护?(小剂量,用于配件清洗)多谢了! 另外MSDS上,他们的闪点都较低,-20C左右,是不是极易燃?多谢了!
作者:http://d.g.wanfangdata.com.cn/Images/head_pic.gif邓芳 http://d.g.wanfangdata.com.cn/Images/head_pic.gif万莉 http://d.g.wanfangdata.com.cn/Images/head_pic.gif崔小平 Author:Deng Fang Wan Li Cui Xiaoping 作者单位:重庆市涪陵区中心医院,重庆,408000 重庆市万州药品检验所,重庆,404000 重庆三峡中心医院,重庆,404000摘要: 建立呋喃西林溶液中呋喃西林含量的高效液相色谱法.方法 色谱柱为Diamonsil C18柱(250 mm x4.6 mm,5μm),流动相为甲醇-水-三乙胺(40:60:0.1),检测波长为254 nm,流速为1.0 mL/min.结果 呋喃西林进样量的线性范围为O...http://ng1.17img.cn/bbsfiles/images/2012/08/201208201647_384759_2379123_3.jpg
化妆品中呋喃香豆素类(三甲沙林、8-甲氧基补骨脂素、5-甲氧基补骨脂素)和欧前胡内酯的检测方法
用微囊藻毒素方法制得的制备液可以用其进行呋喃丹的检测吗?望老师不吝赐教
一把透明的塑料尺子,不小心滴上四氢呋喃,变得不透明了,表面也像被腐蚀了一样,这是什么原因?又试了纯甲醇,也有类似现象……欢迎大家讨论
交流硝基呋喃前处理心得 和经验
请问霍尼韦尔的四氢呋喃多少钱一瓶算合适呢,4L装的。问了好多家差距挺大,大家买的都多少钱呢,在哪买的,谢谢
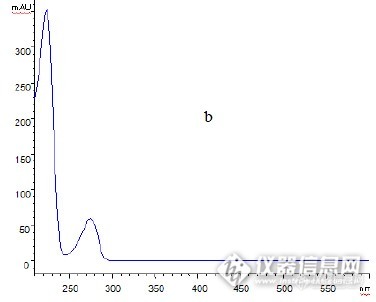
HPLC-DAD分析酸浆中木犀草素及木犀草素-7-β-D-葡萄糖甙成分酸浆(拉丁文名:Physali alkekengi L.)又名红菇娘、挂金灯、戈力、灯笼草、灯笼果、洛神珠、泡泡草、鬼灯等北方称为菇蔫儿、姑娘儿,以果实供食用。化学成分含酸浆苦素A(Physalin A)、酸浆苦素B、酸浆苦素C、木犀草素(Luteolin)及木犀草素-7-β-D-葡萄糖甙。果实含枸橼酸、草酸、维生素C、酸浆红色素(physalien)、酸浆醇(physanol)A,B。花萼含α胡萝卜素、酸浆黄质(physoxanthin)及叶黄素等,种子油的不皂化物中分得多种4α-甲基甾醇,主要为禾本甾醇(gramisterol)和钝叶醇(obtusifoliol)及4种新甾体。此外尚含多种4-脱甲基甾醇,如胆甾醇和24-乙基胆甾醇等。还含有多种三萜3β-一元醇,其中环木菠萝烷醇(cycloartanol)35%,环木菠萝烯醇(cycloartenol)27%、羊毛脂-8-烯-3β-醇(lanost-8-en-3β-ol)。木犀草素(luteolin)是一种天然黄酮类化合物,存在于多种植物中,具有抗炎、抗肿瘤、抗过敏等方面的作用。化学是如下:http://ng1.17img.cn/bbsfiles/images/2016/08/201608311303_607620_2217446_3.jpg目前,国内传统中药有效成分的提取方法普遍存在提取率低、杂质清除率不高、生产周期过长、能耗高、溶剂用量大等缺点。随着中药现代化进程的不断深入,许多现代高新技术不断地被应用到中药有效成分的提取和分离,使得中药有效成分的提取更高效和简便。超声-微波协同萃取技术直接将超声振动与开放式微波两种作用方式相结合,充分利用超声波振动的空化作用以及微波的高能作用,实现了低温常压条件环境下,对固体样品进行快速、高效、可靠的预处理,与常规提取方法相比,超声-微波协同萃取技术具有快速、节能、节省溶剂、污染小等优点。本实验应用超声-微波协同萃取法提取酸浆中的木犀草素及木犀草素-7-β-D-葡萄糖甙,采用高效液相-二极管阵列检测法(HPLC-DAD)测定提取物中木犀草素及木犀草素-7-β-D-葡萄糖甙的含量,药材中二者成分的含量分别为:1.200mg/g 和0.43mg/g,二个峰,木犀草素-7-β-D-葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素-7-β-D-葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,天麻素二个峰位置都发生了蓝移,样品中二个峰的光谱图与标准品二个峰的光谱图相同,可以进一步确定酸浆中含有木犀草素及木犀草素-7-β-D-葡萄糖甙。主要仪器与试剂主要仪器Agilent1100型四元梯度高效液相色谱仪(美国 Agilent 公司)Agilent TC-C18(ODS)色谱柱(5μm,4.6×250mm,美国 Agilent 公司)CW-2000 超声-微波协同萃取仪(新拓微波溶样测试技术有限公司)DJ-10A 型倾倒式粉碎机(上海隆拓仪器设备有限公司)RE-52AA 型旋转蒸发仪(河南巩义仪器厂)LXJ-IIB 型低速大容量多管离心机(上海安亭科学仪器厂)试剂木犀草素(中检所,含量98%;)木犀草素-7-β-D-葡萄糖甙(中检所,含量98%;)酸浆全草(采于黑龙江)除甲醇、乙腈为色谱纯(国药集团化学试剂有限公司),其余试剂除专门提到外,均为分析醇,实验用水为二次蒸馏水。实验方法供试品溶液的制备 精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,加入50mL70%甲醇,开启超声微波,控制在恒温50℃下提取40min,萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm 的微孔滤膜,取续滤液,即得。提取条件的考察溶剂的选择:精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取40min(n=3),萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm的微孔滤膜,取续滤液,HPLC 测定萃取率。溶剂体积分数的选择:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),方法同上。溶剂用量的选择:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,方法同上。提取时间的选择:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),方法同上。提取温度的选择:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,方法同上。对照品溶液的制备 分别精密称取常温减压干燥12h 的木犀草素及木犀草素-7-β-D-葡萄糖甙对照品适量,加甲醇配制成木犀草素-7-β-D-葡萄糖甙为200μg/mL、木犀草素为100μg/mL 的混合对照品溶液,冷藏备用。色谱条件 色谱柱:Agilent TC-C18柱(5μm,4.6×250mm);流动相:A-0.1%乙酸水溶液;B-甲醇,线性梯度洗脱:0~30 min,3%~5% B;30~35 min,5%~20%B;35~40min,20%~20%B;检测波长:270nm;流速:1mL/min;柱温:30℃;进样量:20μL。结果与讨论提取条件的优化结果溶剂的优化结果:分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取30min(n=3),结果表明70%甲醇提取木犀草素-7-β-D-葡萄糖甙的量较高,而木犀草素的量差异不明显,因此选择70%甲醇提取。溶剂体积分数的优化结果:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),结果表明,在甲醇体积分数70%时,木犀草素-7-β-D-葡萄糖甙和木犀草素的提取率随着甲醇浓度的增加而增加;但当甲醇体积分数在70%以上时,木犀草素葡萄糖甙的提取率呈现下降趋势,木犀草素没有明显的变化。木犀草素葡萄糖甙属于一种苷,分子量小,极性较大,当甲醇体积分数过高时,溶液极性降低,使得极性较强的木犀草素葡萄糖甙不易溶出,而木犀草素极性相对木犀草素葡萄糖甙小,影响不明显,因此实验选择70%甲醇作为提取溶剂。溶剂用量的优化结果:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,结果表明溶剂体积在50mL时木犀草素葡萄糖甙和木犀草素的提取率最高,之后随着溶剂用量的增加,木犀草素葡萄糖甙和木犀草素的提取率趋于稳定,因此溶剂用量选用50mL 进行提取 。提取时间的优化结果:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),结果表明超声-微波协同萃取时间从20~40min的过程中木犀草素葡萄糖甙和木犀草素的提取率逐渐增加;而提取时间超过40min之后,提取率反而逐渐下降。超声-微波协同萃取时间太长,植物中大量细胞细胞破碎,使得大量粘性物质等进入提取液,溶剂杂质增多、粘度增大,影响了有效成分的溶出,有效成分含量反而减少,因此选择提取时间为40min。提取温度的优化结果:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,实验表明,提取温度在50~60℃的范围内,木犀草素葡萄糖甙和木犀草素的提取率没有明显差异,考虑到温度太高容易破坏活性成分,因此选择提取温度为50℃。流动相的考察在实验过程中,流动相首先考察了甲醇-水、乙腈-水等度洗脱对酸浆超声-微波协同萃取样品溶液进行分离,乙腈-水作为流动相时,出峰较快,不能较好地把木犀草素葡萄糖甙和木犀草素与其他杂质成分分离;甲醇-水作为流动相时,出现峰形拖尾现象,分离效果不理想。为改善上述现象,改用0.1%乙酸代替水并采用梯度洗脱,经过反复筛选之后,最终确定流动相组成为 A -0.1%乙酸水溶液, B -甲醇,洗脱程序为0~30 min , 3%~5% B;30~35 min ,5%~20% B ;35~40 min 20%~3% B,木犀草素葡萄糖甙和木犀草素和其他杂质成分能够很好的分离,得到较理想的色谱图。对照品溶液和酸浆萃取样品的HPLC-DAD 分析下图分别显示了在上述的色谱条件下,采用 DAD 进行检测得到的两种混合对照品及酸浆萃取样品的 HPLC 分离色谱图。图1色谱图中木犀草素葡萄糖甙和木犀草素的保留时间分别为18.74min, 26.87min,根据保留时间判断,图2中的 a、b 色谱峰分别初步鉴定为木犀草素葡萄糖甙和木犀草素。图3、4分别显示了混合对照品和酸浆萃取物中保留时间18.74min, 26.87min 的色谱峰进行 DAD 检测后得到的光谱图,木犀草素葡萄糖甙和木犀草素 UV 光谱图形状相似,出现 二个峰,木犀草素葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,木犀草素葡萄糖甙二个峰位置都发生了蓝移,样品中二个峰的光谱图与







 我要推广仪器
我要推广仪器
 下载APP
下载APP