测果糖中的二氨基苯乙酮,标准曲线好做吗?为什么我的老是不出峰,且基线不是很稳.紫外220nm,1.0ml/min,柱子C1840度.进样量是300ul很大的,各位有谁做过,给支个招吧谢谢
俺想用氨基苯乙酮做显色剂,不知道有人用过吗?还存在邻,间,对的问题,不知道大家用的哪个?
[color=#444444]用液相检测邻氨基苯乙酮时 前处理为什么要在100摄氏度加热5分钟,然后冷却到室温后再检测呢?[/color]
间硝基苯乙酮、CAS:121-89-1 质量标准 化学试剂 工业级均可,请大家帮帮忙。
新手小白,刚接触GC-MS/MS,我用的是安捷伦7890A-7000B,柱子是DB-5的柱子,走的分别是羟基苯甲酸丁酯和2-氨基苯乙酮的标品,都是1ppm的,溶剂都是丙酮,升温条件50℃——220℃(15℃/min),不分流,然后不出峰。请各位大神多多指导!!!
[color=#444444]最近在做苯乙酮和1-苯乙醇的化学实验,想用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行分析,结果发现两个的沸点相差很近,请问这种情况下应该怎么样进行测试,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的操作应该注意什么,苯乙酮的沸点是202.3度,1-苯乙醇的是203.4度,谢谢大家[/color]
色谱柱DB23和DB-5都试过,都没有标准品峰进样口温度230,检测器温度250,柱温:60℃,以10℃/min程序升温至120℃,恒温1min,以10℃/min程序升温至200℃,恒温2min,进样量1uL;标准品为1uL苯乙醇溶于2ml乙酸乙酯溶剂峰正常,相同条件下苯乙酮标准品也有峰。为什么1-苯乙醇标准品不出峰???
气相色谱仪如何测定苯乙酮的含量,我没学过很想知道,我们厂生产苯乙酮但是不知道怎么用色谱仪做,测定含量
问一下高手们,氨基苯乙酮的含量高低与温度的高低有一定的比例关系吗?如果液体的话能用活性炭给他除去一些吗?
有关苯乙酮的性质如下外观与性状:无色或淡黄色低熔点、低挥发性、有水果香味的固体。熔点(℃):19.7,相对密度(水=1):1.03(20℃),沸点(℃):202.3这么低熔点的物质在室温下(如现在20°左右)基本都是液体了,那么怎么配置30ug/ml的甲醇溶液呢?如何称量?如果用量筒,又怎么计算啊?
请问专家能否告诉我对氯苯乙酮的大概分析方法?
哪位同志有对羟基苯乙酮的滴定含量检测方法.我这里有一个方法,但总是滴不好,用甲醇钠溶液进行滴定,用二甲基甲酰胺进行溶解,用麝香草酚蓝作指示剂.目前存在的问题就是终点变化不明显.哪位有用到或碰到类似情况,都来说说吧.
据外媒报道,近日部分德国媒体报道称,珍珠奶茶含有多种致癌成分,严重危害人体健康。此消息一出,德国珍珠奶茶专卖店的生意变得异常惨淡,部分店家甚至面临关门歇业的境地。 如何检测苯乙酮、溴化物及苯乙烯等致癌物?
[color=#444444]我在安捷伦[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]上(色谱柱是HP-5)分别进了苯乙酮和α-苯乙醇,两个物质出峰时间差不多,然后我又进了二者的混合物(体积比1:1),结果两峰重叠在一起,我调试了柱温,分流比,都没什么进展。(柱温120,进样温度220,检测器220,分流比60:1),请高手指点一下,是不是色谱柱的问题,还是什么?[/color]
最近要做这两个物质的检测, 不知道有没有老师在做的给点意见和相关资料,谢谢苯乙酮 98-86-2 2-苯基-2-丙醇 617-94-7
我们是用WAX柱做苯乙酮,甲醇超声萃取,但有一个样品用GC-MS做有值,质谱、保留时间一致,但是用LC-DAD走样不出峰,标样有出峰,样品加标也有出峰,请问有人知道是什么原因吗?改如何判定?
氧化器主要含异丙苯,测定其中所含杂质含量,主要杂质有二甲基苯甲醇,苯乙酮和过氧化氢异丙苯等,选用何种色谱柱?如何选择操作条件,我们要使用安捷伦1200液相色谱仪,
问题:请教大家 你们乙醇定量是用什么做内标呀?我们现在用苯乙酮 味道太大
我们是用WAX柱做苯乙酮,甲醇超声萃取,但有一个样品用GC-MS做有值,质谱、保留时间一致,但是用LC-MS走样不出峰,标样有出峰,样品加标也有出峰,请问有人知道是什么原因吗?改如何判定?
这几天用岛津的碳18分析氨基酸,用的异硫氰酸苯酯为衍生剂。 用水代替氨基酸标准品,会出现很多杂峰,所以想应该是衍生过程引进的。衍生的方法是 标准品+三乙胺乙腈溶液+异硫氰酸苯酯乙腈溶液 反应一个小时候用正己烷萃取各位大虾有更好的衍生方法么?本实验室没有氮吹仪,没法吹干哦
我们需做苯乙酮还原成苯乙醇的反应无副反应,想测其转化率。以前都是用的是面积归一法做,但他想用更精确点的办法解决,请问除了内标法外,是不是可以用测出那两种物质的响应因子来解决?还是用面积归一法就可以了?测响应因子该怎么测?
大家有接触过盐酸联苯胺或其它芳香胺的盐酸盐吗?比如,盐酸联苯胺,4-氨基偶氮苯盐酸盐,等。这些用在纺织品的用途是什么?对人体有什么危害?有标准或技术方法可以检测吗?
原料药盐酸米多君标准(国内:YBH09452006),5-氨基乙酰丙酸盐酸盐标准。若有特殊要求也可站内信息联系!有国外标准也行。
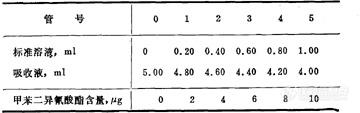
空气中甲苯二异氰酸酯的测定方法 盐酸萘乙二胺比色法 1 原理甲苯二异氰酸酯水解产生相应的芳香胺,芳香胺与亚硝酸钠重氮化后,再与盐酸萘乙二胺偶合生成紫红色,比色定量。2 仪器2.1 冲击式吸收管。2.2 抽气机。2.3 流量计,0~1L/min。2.4 具塞比色管,25ml。2.5 恒温水浴箱。2.6 分光光度计。3 试剂3.1 吸收液:量取25ml盐酸(20=1.19g/ml)与500ml水混匀,加入250ml二甲基甲酰胺,用水稀释成1L,临用前配制。3.2 亚硝酸钠-溴化钠溶液:称取3g亚硝酸钠和5g溴化钠,用80ml水溶解后,稀释成100ml。3.3 氨基磺酸铵溶液,100g/L。3.4 盐酸萘乙二胺溶液,10g/L。3.5 标准溶液:于25ml量瓶中加入5ml二甲基甲酰胺,准确称量,加入1~2滴甲苯二异氰酸酯,再准确称量,两次称量之差即为甲苯二异氰酸酯的质量,然后用二甲基甲酰胺稀释到刻度。计算1ml溶液中甲苯二异氰酸酯的含量,再用二甲基甲酰胺稀释成1ml=100微克甲苯二异氰酸酯的溶液。取5.0ml上述溶液于50ml量瓶中,加入7.50ml二甲基甲酰胺及1.25ml盐酸,混匀后用水稀释至刻度,配成1ml=10微克甲苯二异氰酸酯的标准溶液。4 采样将装有10ml吸收液的冲击式吸收管以1L/min的速度抽取50L空气。5 分析步骤5.1 对照试验:同采样,将吸收管装好吸收液带至现场,但不抽取空气,照样品分析。5.2 样品处理:采样后,用吸收管中的吸收液洗涤进气管内壁3次,由每个吸收管中各量取5.0ml样品溶液,分别放入比色管中,供测定用。5.3 标准曲线的绘制:按表2配制标准管。表2 甲苯二异氰酸酯标准管的配制[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201410_52373_1625938_3.jpg[/img]向各标准管中各加入0.2ml亚硝酸钠-溴化钠溶液(3.2)混匀,放置1min,再加0.2ml氨基磺酸铵溶液(3.3),激烈振摇,待气泡消失后,放置2min,加入1ml盐酸萘乙二胺溶液(3.4),充分混匀后,在20℃水浴中,放置20min,于波长560nm下比色,以甲苯二异氰酸酯含量对吸光度作图,绘制标准曲线。5.4 测定:按5.3相同的操作条件,将处理后的样品进行测定,由标准曲线上查出甲苯二异氰酸酯含量。6 计算X=2C/V0式中:X——空气中甲苯二异氰酸酯的浓度,mg/m3;C——吸收管中所取样品溶液中甲苯二异氰酸酯的含量,?g;V0——标准状况下的样品体积,L。7 说明7.1 本法的检测限为0.9?g/5ml。测定范围为2~10微克/5ml。当甲苯二异氰酸酯的浓度为2、4、6、8、10微克/5ml时,变异系数分别为7.1%、5.8%、6.5%、5.6%、5.8%。7.2 采样后样品可保存3天。7.3 吸收液应临用前配制,因久置后其中盐酸逐渐与二甲基甲酰胺作用而失去酸性。
GB 5009.33-2010中, 对氨基苯磺酸溶液(4 g/L):称取0.4g对氨基苯磺酸,溶于100 mL 20 %(V/V)盐酸中,置棕色瓶中混匀,避光保存。而GB/T 5009.33-20008中 对氨基苯磺酸溶液(4 g/L):称取0.4g对氨基苯磺酸,溶于100 mL 20 %盐酸中,置棕色瓶中混匀,避光保存。我们在使用2008版的标准时是按照盐酸的质量分数来计算的,即若配制500ml20%盐酸(浓盐酸按37%质量分数)需浓盐酸270ml,2010版标准是20%ml浓盐酸加80%ml水混合,盐酸的作用只是制造弱酸环境,酸性变弱了,测了一下发现相比新国标结果值小了1mg/kg左右,这是怎么回事呢?
[font=宋体][size=14px][/size][/font][font='微软雅黑','sans-serif']盐酸中甲苯测定,可以直接萃取后进样分析吗?标准样品配制需要在盐酸中加入不同量的甲苯配制吗?[/font][font='微软雅黑','sans-serif']求助问题来自微信群。[/font][font=宋体][size=14px][/size][/font]
哪位朋友知道 Tg标准品-聚苯乙烯的标准品哪里买啊?
各位XDJM,玻璃化转变温度标准品--聚苯乙烯哪有可以买啊??GB上有的!
如题!苯乙醇胺A 的标准品哪里能买到啊?
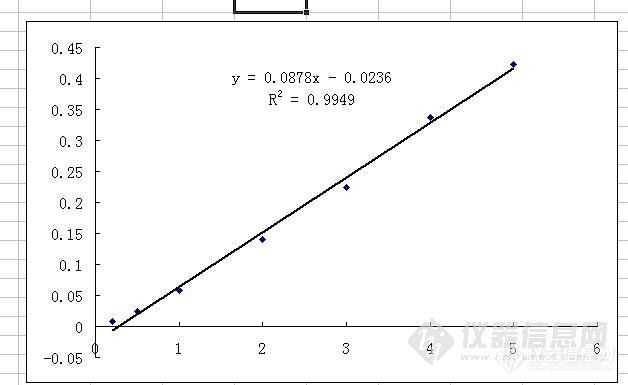
本人做了4天氰化物标准曲线了,采用的是HJ 484-2009异烟酸吡唑啉酮分光光度法,4次做出来的标准曲线都是中间几个点吸光度值明显偏低,重新配制过试剂,也用过30℃水浴锅恒温,还做过人员比对,做出来结果基本都是中间几个点吸光度值明显偏低,今天的测试结果为:空白0.015,扣除空白后0.2ug点吸光度0.008,0.5ug点吸光度0.024,1.0ug点吸光度0.058,2.0ug点吸光度0.140,3.0ug点吸光度0.224,4.0ug点吸光度0.337,5.0ug点吸光度0.423,今天测的空白有点高,但是前3天空白都是0.004和0.005。有没有做过异烟酸吡唑啉酮法测氰化物标准曲线的大虾,帮忙分析下原因,急啊!先谢谢了!http://ng1.17img.cn/bbsfiles/images/2010/11/201011201038_260810_1644065_3.jpg俺帮您做了一个曲线,这样大家看着方便点(二虎)







 我要推广仪器
我要推广仪器
 下载APP
下载APP