一阶导数分光光度法测定盐酸普鲁卡因溶液的含量刘素琴(江苏省金坛市人民医院,江苏 金坛 213200)联系电话:0519-2266680 E-mail:Liusuqin666@163.com 文章编号:04040399摘要 目的:改进盐酸普鲁卡因溶液的含量测定方法。 方法:以一阶导数光谱在308.0nm波长处谷—零间的振幅为定量依据,测定盐酸普鲁卡因溶液的含量。结果:盐酸普鲁卡因溶液浓度在5~30μg/mL范围内与一阶导数谷—零间的振幅呈良好的线性关系,r=0.9999,平均回收率为100.04%,RSD=0.43%。结论:方法简便、准确,可用于测定盐酸普鲁卡因溶液的含量。关键词 一阶导数; 分光光度法; 盐酸普鲁卡因 ;含量[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=32338]一阶导数分光光度法测定盐酸普鲁卡因溶液的含量[/url]
各位:谁有乙酰苯胺的标准谱图呢?我这里有个样品,可能是乙酰苯胺,但是感觉不像,想请大家帮忙给我传一张!!谢谢!
GB/T 5009.140-2003 饮料中乙酰磺胺酸钾的测定标准是饮料中的,其他产品用这个标准该怎么做?比如糕点是否这个标准就不适用于除饮料外的其他产品?大家在使用这个标准的时候如何应用的呢?
食品中甲胺磷和乙酰甲胺磷农药残留量的测定方法1.适用范围本方法适用于谷物、蔬菜和植物油中甲胺磷和乙酰甲胺磷的残留量分析,其最小检出限分别为7.79×10-12g和1.79×10-11g。2.原理概要含有机磷的样品在富氢焰上燃烧,以HPO碎片的形式,放射出波长526nm的特征光,这种特征光通过滤光片选择后,由光电倍增管接收,转换成电信号,经微电流放大器放大后,被记录下来,样品的峰高与标准品的峰高相比,计算出样品相当的含量。3.主要试剂和仪器3.1.主要试剂丙酮;二氯甲烷:重蒸;无水硫酸钠;活性炭:用3mol/L盐酸浸泡过夜,抽滤,用水洗至中性,在120℃下烘干备用;甲胺磷(methamidophos):≥99%;乙酰甲胺磷(acephate):≥99%;甲胺磷和乙酰甲胺磷标准溶液的配制:分别准确称取甲胺磷和乙酰甲胺磷的标准品,用丙酮分别制成0.1mg/mL的标准储备液。使用时用丙酮稀释配制成单一品种的标准使用液(1mg/mL)和混合标准工作液(每个品种浓度为1mg/mL)。贮藏于冰箱中。3.2.仪器气相色谱仪:具有火焰光度检测器;电动振荡器;K-D浓缩器或旋转蒸发器;离心机。4.试样的制备取谷物实验样品经粉碎机粉碎,过20目筛后,制成谷物试样。取蔬菜实验样品洗净,晾干,去掉非食部分后剁碎或经组织捣碎机捣碎,制成蔬菜试样。5.过程简述5.1.提取和净化蔬菜:称取蔬菜试样10g,精确至0.001g,用无水硫酸钠(因蔬菜含水量不同而加入量不同,约50~80g)研磨呈干粉状,倒入具塞锥形瓶中,加入0.2~0.4g活性炭(根据蔬菜色素含量)及80mL丙酮,振摇0.5h,抽滤,滤液浓缩定容至5mL,待气相色谱分析。谷物:称取谷物试样10g,精确至0.001g,置于具塞锥形瓶中,加入40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。小麦:称取小麦试样10g,精确至0.001g,置于具塞锥形瓶中,加入0.2g活性炭及40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。植物油:称取植物油试样5g,用45mL丙酮分次洗入50mL的离心管内,加入5mL水,混匀,在3 000r/min下离心5min,吸取上清液,下面油层再加10mL水和10mL丙酮,离心5min,吸取上清液,合并两次上清液,用K-D浓缩器浓缩近干,残渣和水加入40g无水硫酸钠,研磨呈干粉状,倒入具塞锥形瓶中,加入0.3g活性炭、60mL二氯甲烷,振荡0.5h,抽滤,定容至5mL,待气相色谱分析。5.2.色谱条件色谱柱:玻璃柱,内径3mm,长0.5m,内装2%dEGS/Chromosorb W AWdMCS,80~100mesh。气流:载气,氮气70mL/min,空气0.7kg/cm2,氢气1.2kg/cm2。温度:进样口200℃,柱温180℃。5.3.测定定性:以甲胺磷和乙酰甲胺磷农药标样的保留时间定性。定量:用外标法定量,以甲胺磷和乙酰甲胺磷农药已知浓度的标准样品溶液作外标物,按峰高定量。6.结果计算Xi=hi•Esi•V1hsi•V2•m式中:Xi——样品中i组分有机磷含量,mg/kg;Esi——注入标样中i组分有机磷的含量,ng;hi——样品的峰高,mm;hsi——标样中i组分的峰高,mm;V1——浓缩定容体积,mL;V2——注入色谱样品的体积,μL;m——样品的质量,g。7.方法的精密度添加回收试验中甲胺磷和乙酰甲胺磷的变异系数分别为2.36%和3.95%。8.甲胺磷和乙酰甲胺磷的保留时间在5.2的气相色谱条件下,甲胺磷的保留时间为0.9min,乙酰甲胺磷的保留时间为1.9min。9.来源:GB 14876—94
[font=宋体][size=14px][/size][/font][font='微软雅黑','sans-serif']你好,想问一下老师[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]跑乙酰甲胺磷单标的时候不出峰,跑有机磷混标的时候乙酰甲胺磷可以出峰,换新的一支标准品也是同样的情况,想请教一下老师是哪里有问题,谢谢[/font][font='微软雅黑','sans-serif']求助问题来自微信群。[/font][font=宋体][size=14px][/size][/font]
请教各位:元素分析用的标准物质乙酰苯胺的定值不确定度是从哪里得知的?我们实验室里的乙酰苯胺标准物质是从上海市计量测试技术研究院买的,在标准物质证书上只有百分理论值,并没有列出不确定度。
如题:乙酰甲胺磷的标准溶液怎么存放?我们的是用甲醇稀释到50PPM在冰柜保存,6月配标,9月测试时响应挺高的,10月测试发现响应低了很多,仪器条件没变,这是为什么?
[table][tr][td]乙酰磺胺酸钾(又名安赛蜜)的检测标准那位老师哪里有?求分享?谢谢![/td][/tr][/table]
用串接质谱(包括单极)检测乙酰甲胺磷时离子碎片比不稳定,也就是说走标准,离子碎片比例都不一致。为什么?大家有没有发现过。希望专家给予答复。当然19648中也没有乙酰甲胺磷这个目标物。
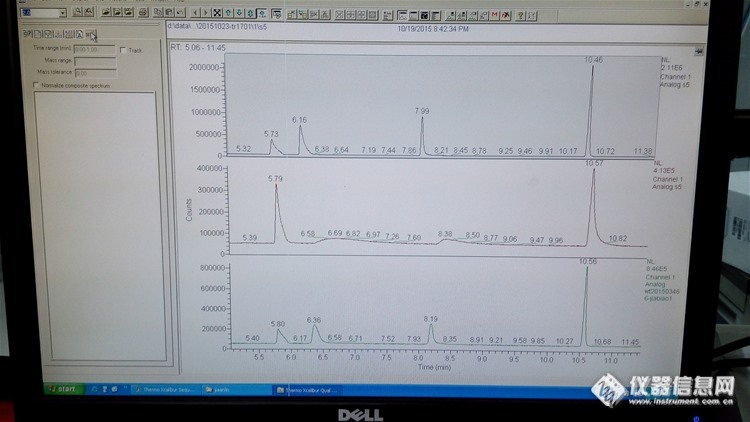
http://ng1.17img.cn/bbsfiles/images/2015/11/201511111107_573060_2547863_3.jpg如图,第一个谱图是1个月前做的标准曲线点,峰分别是敌敌畏,甲胺磷,乙酰甲胺磷,乐果,用的柱子是35柱第二个谱图是前天做的标准曲线点,其中甲胺磷,乙酰甲胺磷峰塌下去了第三个谱图是前天做的红枣加标样品,其中甲胺磷,乙酰甲胺磷峰很正常。如今往红枣样品上机液(四个峰均不出峰)添加混标液,得出的谱图和第三个一样;重新配了混标,甲胺磷和乙酰甲胺磷也是超级拖尾,但是一旦把混标加入样品液里面,两个出峰都很正常求解决方法(已经切割过柱子前端,进样瓶都是新的,除了溶液一个是丙酮定容的样品上机液,一个是色谱纯丙酮不一样外,其他条件都一样)
急求标准对二氯苯、五氯化磷、乙酰氯、对甲苯磺酰胺、乙二醇甲醚、马来酸国标、化工标准、地方标准都行谢谢!![em0808]
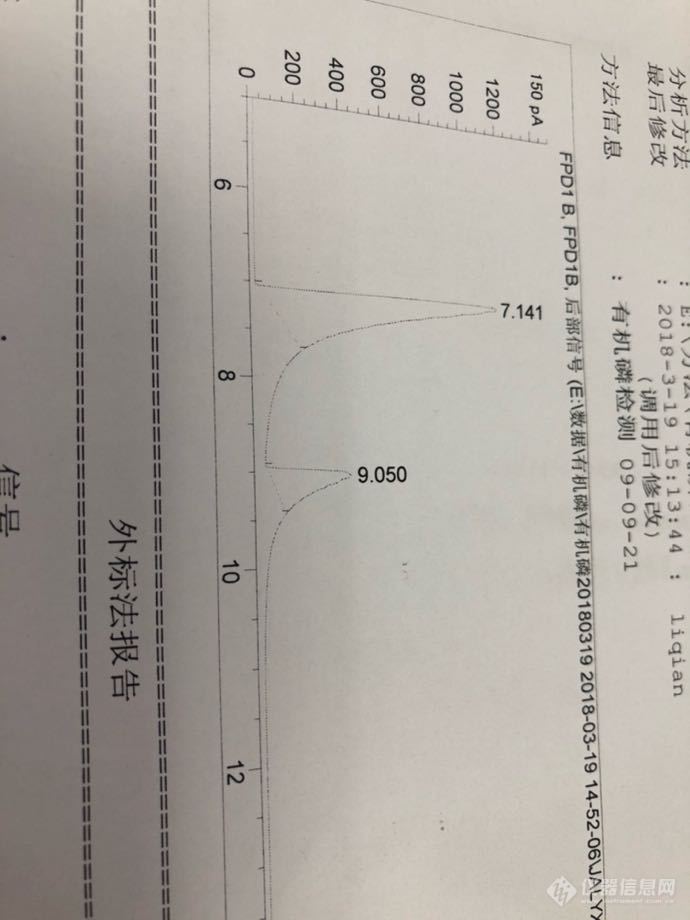
甲胺磷 乙酰甲胺磷拖尾严重…请问有啥解决办法吗?衬管也是新换的,柱头也割过一截了。方法也是之前用的。标准品也是新开的…请问有啥解决办法吗?[img=,690,920]http://ng1.17img.cn/bbsfiles/images/2018/03/201803191602167875_8255_3379628_3.png[/img]
第十章 胺类药物的分析掌握盐酸普鲁卡因、盐酸利多卡因、盐酸丁卡因和对乙酰氨基酚的鉴别、杂质检查和含量测定方法。 掌握肾上腺素、盐酸去氧肾上腺素及其制剂的鉴别、杂质检查和含量测定方法。 第一节 盐酸普鲁卡因的分析有芳伯氨基特性,显重氮化-偶合反应。含酯键易水解,产物主要为对氨基苯甲酸(PABA)。脂烃胺侧链为叔胺氮原子,具有弱碱性。盐酸盐系白色结晶性粉末,具有一定的熔点,易溶于水和乙醇,难溶于有机溶剂。一、鉴别1.重氮化-偶合反应 分子结构中具有芳伯氨基或潜在芳伯氨基的药物,均可发生重氮化反应,生成的重氮盐可与碱性β-萘酚偶合生成有色的偶氮染料。 2.水解反应 取本品约0.1g,加水2ml溶解后,加10%氢氧化钠溶液1ml,即生成白色沉淀,加热变为油状物(普鲁卡因);继续加热,产生的蒸汽(二乙氨基乙醇)能使湿润的红色石蕊试纸变为蓝色;热至油状物消失后(生成可溶于水的对氨基苯甲酸钠),放冷,加酸酸化,即析出白色沉淀。此沉淀能溶于过量的盐酸。3.氯化物反应沉淀反应 在硝酸酸性条件下与硝酸银生成白色沉淀,沉淀加氨试液即溶解。氧化还原反应 加二氧化锰混匀,硫酸润湿,加热产生氯气,能使湿润淀粉碘化钾试纸显蓝色。4.红外光谱法二、特殊杂质检查普鲁卡因分子结构中有酯键,易发生水解反应。其注射液制备过程中受灭菌温度、时间、溶液pH值、贮藏时间以及光线和金属离子等因素的影响,可发生水解反应生成对氨基苯甲酸和二乙氨基醇。其中对氨基苯甲酸随贮藏时间的延长或高温加热,可进一步脱羧转化为苯胺,而苯胺又可被氧化为有色物,使注射液变黄,疗效下降,毒性增加。故中国药典90年版规定,本品注射液应检查水解产物对氨基苯甲酸,其限度不得超过1.2%。检查方法 薄层色谱法。供试品溶液2.5mg/ml与对照品溶液30μg/ml分别点于硅胶H薄层板上,展开后用对二甲氨基苯甲醛溶液喷雾显色。供试品溶液如显与对照品溶液相应的杂质斑点,其颜色与对照品溶液主斑点比较,不得更深。三、含量测定 分子结构中具有芳伯氨基,可用亚硝酸钠滴定法测定含量。
今天发现有些原料药没有用红外鉴别,比如胺类药物:普鲁卡因、利多卡因、对乙酰氨基酚、肾上腺素中,肾上腺素没有使用红外鉴别,为什么?IR的鉴别力不是很强吗?为什么不收入药典?苯巴比妥类药物,苯巴比妥、司可巴比妥钠、硫喷妥钠中,硫喷妥钠没有使用IR鉴别,why?
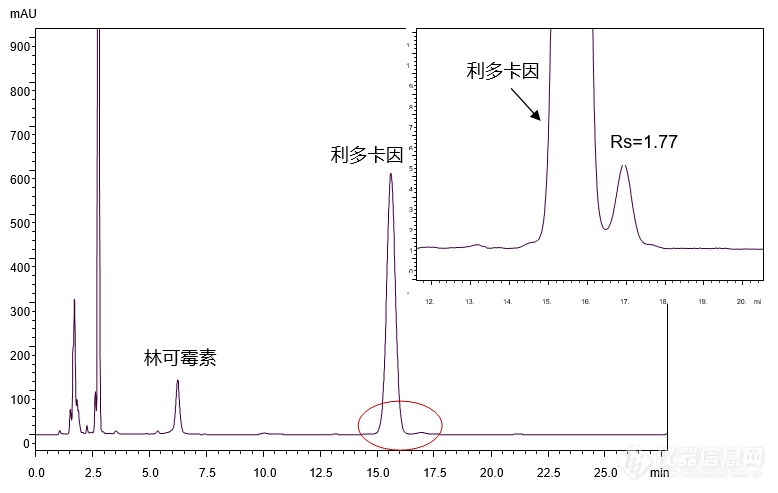
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
最近做绿茶提取物甲胺磷,乙酰甲胺磷,对硫磷的检测,回收率做不出,甲胺磷乙酰甲胺磷才4-5%,对硫磷有70-80%。我使用的是安捷伦的6890N,检测器NPD,不分流进样,进标准品1.0ug/ml峰面积大概是300左右,但是不是很稳定。感觉问题出在前处理,分别用正己烷,乙酸乙酯,乙腈做过过加样回收,加2ml浓度1.0ug/ml。乙酸乙酯,乙腈提取浓缩后为粘稠红棕色液体,用无水硫酸镁和活性炭分散固相萃取后没有改善,正己烷提取后浓缩液无色。浓缩前进过仪器是可以测出来的,浓缩后没有了,回收液液没有,估计分解了。浓缩使用的是步其的平行定量浓缩仪,水浴45度,真空度250Mbar,冷凝水10-13度。我认为是浓缩步骤出的问题,大家帮分析一下,到底是哪里出的问题?有没有跟好的前处理分享一下,要方法简单的哦
我是新手,导师希望我用质谱来测定一下UDP-GlcNAc的含量,全称就是N-乙酰葡糖胺。我曾经用三乙胺醋酸作为流动相,用高效液相测定了含量,出峰时间在8分钟左右当直接标准品进样时,出峰比较快,很正常、但是当用三乙胺醋酸流动相时,标准品仍然用甲醇溶,却检测不到任何峰了这是怎么回事?我是一定要更改流动相了是吗?我查不到有关的流动相的资料,求各位专家大侠帮帮我,我该更换什么流动相比较合适?我换了流动相之后,样品仍然用纯甲醇溶解吗
问:请问乙酰氯的GC分析方法极其标准?我们是是化工企业,生产高纯乙酰氯,分析量很大,我们目前用GC-FID毛细管柱子,没有标准品,含量大约99。5%,请问这样分析的缺陷,用HPLC行不行?假如有其他微量杂质,是否还要用质谱仪?
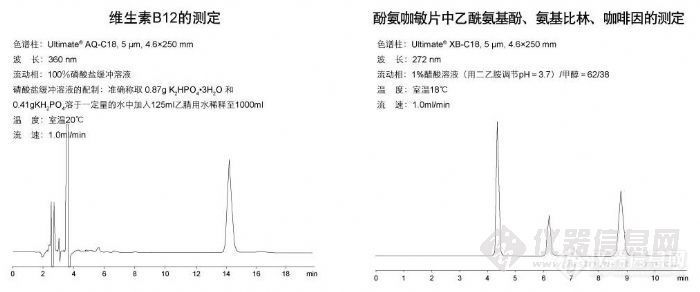
维生素B12的测定和酚氨咖敏片中乙酰氨基酚、氨基比林、咖啡因的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021850_180185_1896702_3.jpg
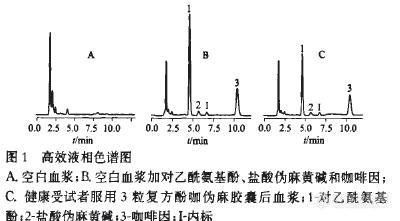
【作者】 胡晓; 张丽芳; 温金华; 蔡军;【机构】 南昌大学医学院临床药理研究所;【摘要】 目的:建立一种同时测定人血浆中对乙酰氨基酚、伪麻黄碱和咖啡因的高效液相色谱(HPLC)法,并用于含上述组分的复方制剂的人体药动学研究。方法:以茶碱为内标,血样经醋酸乙酯提取后,采用高效液相色谱紫外(HPLC-UV)法进行测定。色谱柱为Diamonsil C18柱(4.6mm×150mm,5μm);流动相为甲醇-0.05mol.L-1磷酸二氢钾(23∶77,pH2.4);流速1mL.min-1。检测波长210nm。结果:人血浆中对乙酰氨基酚、盐酸伪麻黄碱和咖啡因质量浓度测定的线性范围分别为0.12~11.52mg.L-1,0.008~0.432mg.L-1和0.03~2.16mg.L-1;最低可定量质量浓度分别为0.12,0.008,0.03mg.L-1;各组分日内、日间RSD均小于15%,方法回收率均大于88%。结论:该方法能快速可靠地同时测定人血浆中对乙酰氨基酚、伪麻黄碱和咖啡因的浓度,可用于含上述组分的复方制剂的人体药动学或生物等效性研究。【关键词】 http://ng1.17img.cn/bbsfiles/images/2012/08/201208142012_383851_1609970_3.jpg
2008年9月22日,日本厚生劳动省发布食安输发第0922003号通知:近日,根据检疫所的监控检查结果,中国产新鲜胡萝卜中查出了乙酰甲胺磷。因此,日本厚生劳动省决定在对中国产胡萝卜及其加工品(限于简单加工)实行的命令检查的检查项目中追加乙酰甲胺磷。 违反事例: 1. 产品名称:新鲜胡萝卜 产 地:中国 申报数量及重量:2500箱,25000kg 检查结果:乙酰甲胺磷 0.02ppm(标准值:0.01ppm) 申报处:东京检疫所 违反确定日期:2008年5月28日 处理状况:全部保管中 2. 产品名称:新鲜胡萝卜 产 地:中国 申报数量及重量:2500箱,25000kg 检查结果:乙酰甲胺磷 0.03ppm(标准值:0.01ppm) 申报处:神户检疫所 违反确定日期:2008年9月19日 处理状况:1975箱已出库,525箱保管中国
4-氯乙酰乙酸乙酯的检测标准及检测方法N,N-二乙基乙二胺、N,N-二甲基乙二胺、N-乙基乙二胺系列产品,质量按Q/320801GHD001-2004标准执行、含量min.99.%
现在手头有两个,一个是《饮料中乙酰磺胺酸钾的测定》GB/T5009.140-2003 另一个是 《食品中的乙酰磺胺酸钾的测定》,用哪个合适呢?用经验的人给指导一下,谢啦~
第一章 麻醉药 第一节 全身麻醉药 1、 吸入麻醉药 氟烷:2-溴-2-氯-1,1,1-三氟乙烷 起效、苏醒快、作用弱,全麻及诱导麻醉 性质:1、氧瓶燃烧 2、加入硫酸,沉于底部。 甲氧氟烷浮于硫酸上层。 甲氧氟烷:麻醉作用和肌松作用比氟烷强,诱导期长。 恩氟烷:新型高效吸入麻醉药,麻醉肌松作用强,起效快,临床常用。 异氟烷为异构体 乙醚:氧化后生成过氧化物对呼吸道有刺激作用。 2、 静脉麻醉药 盐酸氯胺酮:2-(2-氯苯基)-2-(甲氨基)环已酮盐酸盐 2个旋光异构体,用外消旋体 作用快、短、副作用小,诱导期短。 分离麻醉 羟丁酸钠: 作用弱、慢、毒性小。 --OH 1、三氯化铁红色 2、硝酸铈铵橙红色 第二节 局部麻醉药 一、 对氨基苯甲酸酯类 构效关系:1、苯环上增加共他取代基时,因增加空间位阻酯基水解减慢,局麻作用增强。 2、苯环上氨基的烃以烷基取代,增强局麻作用。 丁卡因 3、改变侧链氨基的取代基,有些作用增强。 布他卡因 4、羧酸中的氧原子若以电子等排体硫原子替代(硫卡因),脂溶性增大,作用增强。 盐酸普鲁卡因:4-氨基苯甲酸-2-(二乙氨基)乙酯盐酸盐 不宜表面麻醉 性质:1、加氢氧化钠有油状普鲁卡因析出。 干燥稳定,避光 PH=3-3.5最稳定。 2、酯键:水溶液水解失活:对氨基苯甲酸及二乙氨基乙醇,前者氧化变色 3、叔胺结构:碘、苦味酸等呈色 4、芳伯氨反应: 盐酸丁卡因:4-(丁氨基)苯甲酸-2-(二甲氨基)乙酯盐酸盐 作用:用于粘膜麻醉,与普鲁卡因一起成为应用最广的局麻药。 二、酰胺类: 盐酸利多卡因:N-(2,6-二甲基苯基)-2-(二乙氨基)-乙酰胺盐酸盐-水合物 性质:酰胺键较酯键稳定,酸碱中均较稳定 。作用强,可用于表面麻醉 布比卡因:1-丁基-N-(2,6-二甲苯基)-2-哌啶甲酰胺盐酸盐 长效局麻药,用于浸润麻醉。 三、氨基酮类及氨基醚类
想问问大家 食品中的安赛蜜(乙酰磺胺酸钾)大家都是用什么方法测定的 GB/T5009.140-2003只规定了饮料中的
问题:.请问甲维盐或 乙酰甲胺磷的制剂用什么方法来做?有标准号吗?
关于乙酰氯的国家标准在网上找了很久,找到了份氯乙酰氯的然后发现了乙酰氯GOST 5829-1971乙酰氯 技术条件 (俄罗斯标准)GJB 702-1989 氯乙酰氯 (中国国家军用标准)哪位老师有做过,或者了解乙酰氯国家标准的可否帮下忙,传一份给我,急用,先谢谢大家了
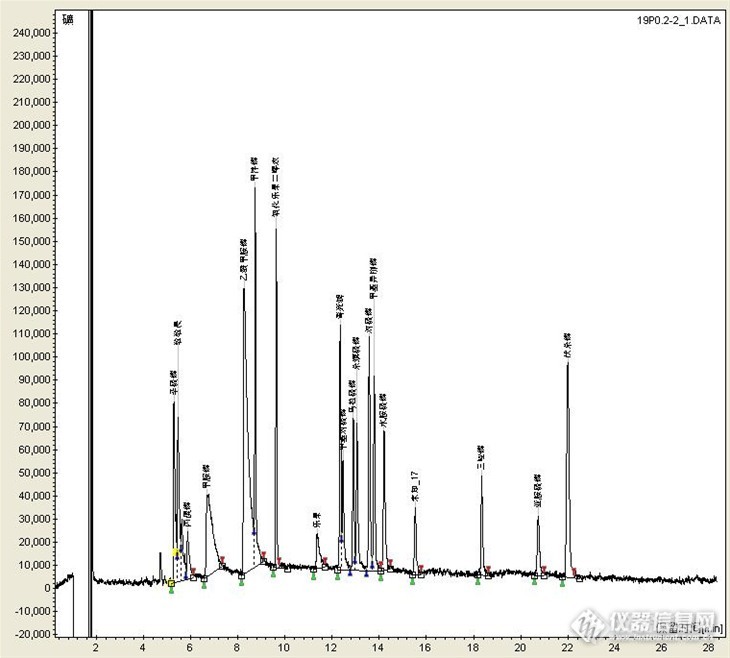
用FPD检测器,DB1701色谱柱做乙酰甲胺磷时,容易被衬管吸附,用高惰性衬管出峰,但峰有拖尾现象,那么有没有人用别的型号的色谱柱做乙酰甲胺磷,检出限低而出峰尖锐?http://ng1.17img.cn/bbsfiles/images/2013/05/201305231601_441223_1645480_3.jpg这是DB1701(30*0.25*0.25)色谱柱的乙酰甲胺磷出峰情况。
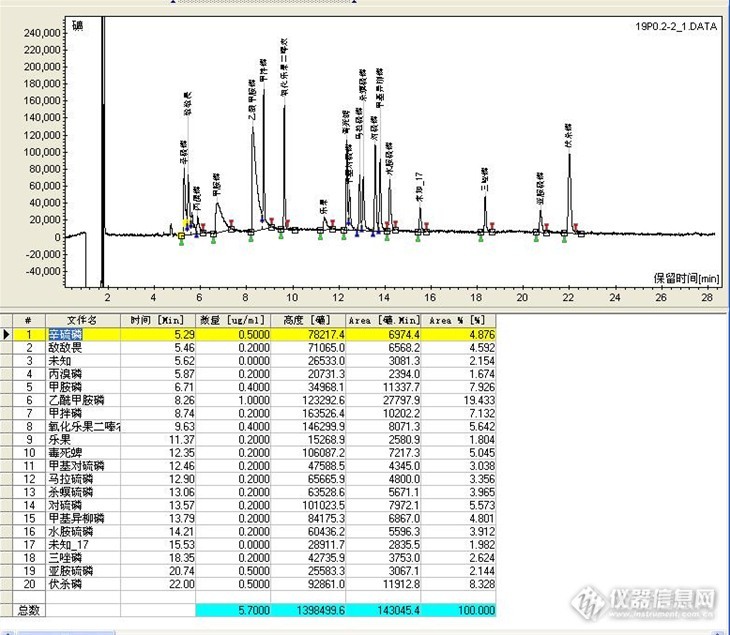
用FPD检测器,DB1701色谱柱做乙酰甲胺磷时,容易被衬管吸附,用高惰性衬管出峰,但峰有拖尾现象,那么有没有人用别的型号的色谱柱做乙酰甲胺磷,检出限低而出峰尖锐?http://ng1.17img.cn/bbsfiles/images/2013/05/201305231603_441225_1645480_3.jpg这是DB1701(30*0.25*0.25)色谱柱的乙酰甲胺磷出峰情况。
有没有人做咖啡因的啊?现在我们公司想买咖啡因标准品,但是问了好几个地方,都说管制的,没法买。想问问大家哪里买的







 我要推广仪器
我要推广仪器
 下载APP
下载APP