摘要:建立胡黄连中香草酸和桂皮酸的含量测定方法。方法用双波长扫描法测定胡黄连中香草酸和桂皮酸的含量。结果香草酸。桂皮酸斑点峰面积3Il内稳定,香草酸回收率为103.86%,RSD=1.33%,桂皮酸回收率为103.16%,RSD=1.28%。结论该方法稳定,可行。具有实用性。 关键词:胡黄连 薄层扫描法 香草酸 桂皮酸 胡黄连具有保肝利胆、抗炎、抗真菌等药理作用。胡黄连含胡黄连素、胡黄连苷(I II III)、D-甘露醇、香草酸、肉桂酸、胡黄连醇成分。香草酸和桂皮酸是其中的两种抗菌成分。我们对胡黄连中香草酸、桂皮酸含量建立了薄层扫描法,以达到控制胡黄连的质量,从而为临床疗效提供保证。 1 仪器与试剂 药材:胡黄连,太原市药材公司;仪器:日本岛津CS--9301PC薄层扫描仪;手提式荧光灯(上海固村电光仪器厂);对照品:香草酸对照品(中国药品生物制品检定所);桂皮酸对照品溶液(省药检所提供e=0.604mg/50ml);硅胶GF254(青岛海洋化工厂)所用试剂均为分析纯。 2 实验条件 2.l 薄层层析条件:分别以石油醚-氯仿-丙酮-冰醋酸(10:4.4:10.1);正己烷-乙醚-冰醋酸(5:5:0.1);正己烷-氯仿-乙醚-冰醋酸(5:3:2:0.1)以及氯仿:甲醇(2:1)展开,多次比较发现正己烷。氯仿-乙醚-冰醋酸(5:3:2:0.4)分离效果好。 2.2 测定波长及主要扫描参数,分别对香草酸,桂皮酸对照品斑点在200nm-370nm扫描,在290nm处有最大吸收,350nm处无吸收,固定350nm为参比波长,290nm为测定波长。
有关单位: 经国家食品药品监督管理局化妆品审评专家委员会审核,拟批准“二甲氧基甲苯基-4-丙基间苯二酚”和“聚甲基丙烯酰基赖氨酸”作为化妆品原料使用。现公开征求意见,请于2011年6月27日前将反馈意见电子版发送至chenzh@sfda.gov.cn。 附件:1.“二甲氧基甲苯基-4-丙基间苯二酚”技术要求 2.“聚甲基丙烯酰基赖氨酸”技术要求 国家食品药品监督管理局食品许可司 二〇一一年六月十五日
小弟做了好几次2-溴,3,3‘-二甲氧基联苯的氢谱,都发现甲基有三重峰,做何解?谢了!
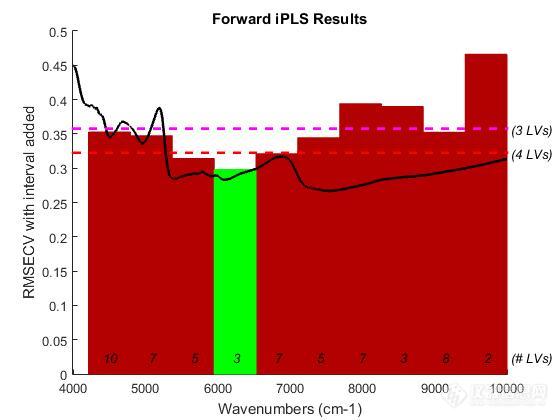
[align=center][b]NIRS用于桂枝中桂皮醛、水分、浸出物含量快速检测方法研究[/b][/align][align=center]研究生:范剑[/align][align=center]导师:臧恒昌教授[/align][b]摘要目的:[/b]干姜和桂枝为传统常用药对。现代药学研究表明,桂枝、干姜均含有大量挥发油且为两药主要药效成分。随着2016年《中药配方颗粒管理办法(征求意见稿)》发布,未来中药配方颗粒限制将逐步放开。相对于单味药材提取的配方颗粒,经典药方或药对形式的配方颗粒,因其更加贴近中医用药理论,将来会受到越来越多的重视。进行干姜和桂枝混合蒸馏提取过程的研究,也可为经典药对配方颗粒的开发提供一定的技术支持。[b]方法:[/b]采用 Antaris II 傅立叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]漫反射模块采集85批桂枝样品[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url],以甲苯法、超高效液相色谱法和浸出物测定法,分别测定样品中水分、桂皮醛和浸出物含量,作为参考值,结合偏最小二乘算法分别建立水分、桂皮醛和浸出物含量的快速定量模型。[b]关键词:[/b]桂枝;[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url];过程分析[align=center]The research on the mixeddistillation extraction of Zingiberis Rhizoma and Cinnamomi Ramulus by NIRS[/align][align=center]Grauate student: Jian Fan[/align][align=center]Supervisor:Hengchang Zang[/align][b]Abstract Objective[/b]:Zingiberis Rhizoma and CinnamomiRamulus are couplet medicinesa in the Traditional Chinese Medicine (TCM). TheZingiberis Rhizoma contains chemical constituents of volatile oil, gingeroletc. It is a common TCM used in medicine and food. Its ether extract and waterextract have obvious analgesic effect. The cassia twig mainly contains cinnamicacid and cinnamaldehyde, it has obvious antipyretic, sedative, antiasthmatic,anti allergic and other effects. TCM on Guizhi - ginger in the compound oftraditional Chinese medicine compatibility is widely used, such as ZhangZhongjing, there are Guizhi drug compatibility in Huang Liantang, smallQinglong Decoction, Chaihuguizhi dried ginger in the “Treatise on FebrileDiseases”. Cassia twig and dried ginger contain a lot of volatile oil, and theyare the main active ingredients of two drugs. Shenzhiling oral solution is onenew kind of traditional chinese drugs , in the production of it,Zingiberis Rhizoma and CinnamomiRamulus as a couplet medicinesa were extracted together in 2016, theregulation of Chinese Medicine Dispensing Granules(take advicing)wes published. In thefuture, the limitations of Chinese Medicine Dispensing Granules will begradually liberalized, the application amount of Chinese Medicine DispensingGranules will be greatly increased. Chinese Medicine Dispensing Granules madewith a classic prescription of Chinese Medicine or couplet medicinesa. In thefuture, more and more attention will be paid to it. Study of ginger and CinnamomiRamulus mixed distilled extraction process, but also can provide technicalsupport for the development of the classic of medicine formula granules. [b]Methods:[/b] Collect 75 near infraredspectroscopys of samples by near-infrared spectrograph with diffuse reflectancemodule. The reference analyses were performed with toluene methodand, UHPLC andpharmacopoeia method respectively for determination of cinnamaldehyde,moisture, and extraction.[b]Key words: [/b]near-infraredspectroscopy manufacture process process analysis techonlogy[b]1 材料与仪器1.1 试剂与样品 [/b]桂皮醛(纯度 98.9 %,批号 110710-201619)购自中国食品药品检定研究院;乙腈、甲醇均为色谱纯;甲苯为分析纯加水饱和后经蒸馏制得;其它等试剂均为分析纯;超纯水(自制);75批桂枝样品购自零售药店、医院药房及药材批发企业,经泰安市食品药品检验检测中心中药科鉴定为樟科植物肉桂的干燥嫩枝。[b]1.2 仪器和软件[/b]Antaris II傅立叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url],PLS_Toolbox工具箱;Agilent 1290型超高效液相色谱仪;Aquity BEH C18 色谱柱;KQ-100DE型医用数控超声波清洗器;电子分析天平; FW80型高速万能粉碎机。[b]2 方法2.1样品制备[/b]将收集的75批桂枝药材粉碎过40目筛,编号,封口袋密封置防潮柜中常温保存,备用。[b]2.2 [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]的采集[/b]取样品粉末约5g,混合均匀后放入样品杯中,摊平,压紧,以空气为参比,扣除背景,采用积分球漫反射方式采集[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]图。光谱扫描范围4000~10000 cm[sup]-1[/sup],分辨率8 cm[sup]-1[/sup],扫描次数32次,每批样品扫描3次,求平均NIR光谱值。[b]2.3 样品中桂皮醛含量的测定[/b](1)对照品溶液的配制精密称取桂皮醛对照品105.00 mg于100 mL容量瓶中,加甲醇溶解并稀释至刻度,再精密量取1 mL至100 mL量瓶中加甲醇稀释至刻度。(2)供试品溶液的制备取桂枝粉末约0.5 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,称定重量,超声处理30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液1 mL ,置25 mL量瓶中,加甲醇至刻度,摇匀,即得。过0. 2 μm 微孔滤膜,供UHPLC分析用。(3)色谱条件WatersAquity BEH C18 色谱柱;流动相水(A)-乙腈(B),梯度洗脱;柱温30 ℃,流速0.3 mL/min,检测波长280 nm,进样体积5 uL。(4)含量测定按照(2)项下供试品溶液配制方法配制各样品供试品溶液,在(3)项的色谱条件下进样分析,利用外标法计算桂皮醛的含量。[b]2.4 样品中水分含量的测定[/b]按照2.2.4项下方法,精密称取样品粉末约15 g,测定计算含量。[b]2.5 样品中浸出物含量的测定[/b] 供试品约2 g,精密称定,置100 mL的锥形瓶中,精密加水50mL,密塞,称定重量,静置1小时后,连接回流冷凝管,加热至沸腾,并保持微沸1小时。放冷后,取下锥形瓶,密塞,再称定重量,用水补足减失的重量,摇匀,用干燥滤器滤过,精密量取滤液25 mL,置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105 ℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量。以干燥品计算供试品中水溶性浸出物的含量(%)。[b]2.6 定量模型的建立[/b]利用化学计量学软件对光谱数据进行处理,建立桂枝中桂皮醛、水分、浸出物含量的PLS定量分析模型。首先,用K-S法按照2:1比例对样品进行校正集和验证集划分;通过光谱预处理方法和建模光谱区间的选择优化建模参数,提高模型稳健性和预测能力。采用模型评价参数 RMSEC、RMSEP、[i]R[sup]2[/sup][sub]c[/sub][/i]、[i]R[sup]2[/sup][sub]P[/sub][/i]、[i]LVs[/i]等参数对模型准确度和预测能力进行评价,并利用配对[i]t[/i]检验对验证集预测结果与测量结果进行显著性检验,进一步评价模型的预测能力。[b]3 结果与讨论3.1 桂皮醛含量结果[/b](1)UHPLC分析方法线性考察UHPLC分析方法线性考察结果:桂皮醛与相邻杂质峰分离度均大于1.5,符合分离度要求,在1.05-21 ug/Ll范围内,标准曲线为y = 166634x + 17.599 ,r[sup]2[/sup] =0.9998,标准曲线线性良好。图3-1为桂皮醛测定中,对照品与样品色谱图。[align=center][img=,690,273]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261509099671_1014_3389662_3.png!w690x273.jpg[/img][/align][align=center][img=,690,273]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261508367711_1478_3389662_3.png!w690x273.jpg[/img][/align]A.对照品;B.样品;[align=center]图3-1 桂枝中桂皮醛含量测定对照及样品的UHPLC[/align](2)桂皮醛含量结果共测定75个样品,其桂皮醛含量范围在0.543 % ~1.83 %。[b]3.2 水分含量结果[/b]共测定75个样品,其水分含量范围在8.38 % ~11.09 %。[b]3.3 浸出物含量结果[/b]共测定75个样品,其水浸出物含量范围在2.09 % ~7.72 %。[b]3.4 [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]定量分析模型的建立3.4.1样品原始光谱图[/b][align=center][img=,544,268]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261510094401_9199_3389662_3.png!w544x268.jpg[/img][/align][align=center]图3-2 桂枝样品的近红外原始光谱叠加[/align]图3-2为不同批次桂枝样品间的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]图,谱图较为相似,近红外原始光谱图与桂皮醛、水分、浸出物含量数据的相关性不显著,故须经过数学处理提取特征信息后,才能建立准确可靠的含量预测模型。[b]3.4.2样品校正集和验证集划分结果[/b]K-S法按照2:1比例对样品进行校正集和验证集划分,选择50个样品用于建立测定桂枝样品中桂皮醛、水分、浸出物含量的定量校正模型,选择25个样品作为验证集,用于验证所建立校正模型的预测能力。校正集和验证集中桂皮醛、水分、浸出物的最大值、最小值和平均值见表3-1。水分、浸出物含量验证集样品包含在校正集中,划分结果可行,有利于建立稳定可靠的模型。[align=center][img=,559,177]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261510500963_8217_3389662_3.png!w559x177.jpg[/img][/align]K-S划分结果,是桂皮醛含量验证集范围超出了校正集,所以用TQ软件自带功能重新对桂皮醛含量模型进行校正集和验证集划分,划分结果见表3-2。[align=center][img=,549,181]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261511341051_7951_3389662_3.png!w549x181.jpg[/img][/align][b]3.3.3桂皮醛、水分、浸出物含量分析模型建立(1)桂皮醛定量分析模型建立[/b]采用TQ Analyst 9. 1 软件自带化学计量学工具对[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]进行预处理,消除固体样本颗粒、光散射、杂散光、仪器响应、以及一些与待测样品性质无关的因素所导致的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]的基线漂移、噪声等。考察未处理(None),S-G平滑,ND平滑,一阶导数(FD),二阶导数(SD),多元散射校正(MSC),标准正态变量变换(SNV)以及其组合的预处理方式。桂皮醛其结构式见图3-3:[align=center][img=,354,472]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261512083981_5013_3389662_3.png!w354x472.jpg[/img][/align][align=center]图3-3 桂皮醛结构式[/align]含苯环,为芳烃化合物,芳烃的一级倍频和二级倍频分别在1685 nm(5934 cm[sup]-1[/sup])和1143 nm(8749 cm[sup]-1[/sup]),组合频在2150 nm(4651 cm[sup]-1[/sup])和2460 nm(4065 cm[sup]-1[/sup])[sup][/sup]。因此,尝试通过手动方法选择不同波段优选建模波段;采用PLS法建立桂皮醛定量校正模型,以校正集样品的以RMSECV、[i]R[sup]2[/sup]c、[/i]RMSEP、[i]R[sup]2[/sup]p[/i]、LVs、Perfformance Index(PI)为指标,优化建模参数。同过桂皮醛定量模型不同光谱预处理方法的分析,可知:同过PI指数可以看出,MSC处理光谱的效果不如原始光谱、SNV处理光谱的效果优于原始光谱、单独微分处理效果均不如原始光谱,二阶导数效果比一阶更差;MSC、SNV分别与FD、SD组合处理光谱效果均有所提升,与FD的组合模型优化效果更明显;当在此组合基础上再加上平滑处理时建模效果反而下降,说明,平滑的过程可能将有效信息掩盖。最佳光谱预处理组合为:MSC+FD、SNV+FD。光谱经预处理后建模评价参数基本接近,仅有细微差别。因此,暂时将两种处理方式均作为最优预处理方式对待。进行下一步的特征波长优化。表3-4是MSC+FD、SNV+FD两种预处理方式与不同光谱波段的建模效果汇总表。从表3-7数据可以看出在用包含芳烃特征吸收的谱段进行建模并没有取得预期的效果,可能与所选取波段不够精准有关系;也可能选取波段使信息量减少,造成了有效信息的丢失;综合考虑MSC+FD、SNV+FD预处理所建模型评价参数认为SNV+FD更优。因此,选择SNV+FD预处理方式,全光谱建立PLS最佳模型,模型参数为[i]R[sup]2[/sup]c[/i]=0.9855,[i]R[sup]2[/sup]p[/i]=0.9601,RMSEC=0.0427,RMSEP=0.0487,LVs为5。[align=center][img=,645,244]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261512437551_5597_3389662_3.png!w645x244.jpg[/img][/align][align=center]图3-4为桂皮醛预测值与实测值相关图[/align]以PLS法建立的最佳模型计算得到的验证集样品的桂皮醛预测值和UHPLC法测定的结果进行配对t检验,以评价模型的预测能力。表3-3为配对t检验的统计学结果,可见UHPLC测定结果的平均值和NIRS得到的结果均值相同。在95%的置信限下,桂皮醛模型的P=0.4510.05,说明近红外模型预测的结果和UHPLC的测定结果没有显著性差异,证实了NIRS用于桂枝药材桂皮醛测定的有效性。[align=center][img=,575,160]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261513515655_413_3389662_3.png!w575x160.jpg[/img][/align][align=center][/align][b](2)水分定量分析模型建立[/b]用Matlab化学计量学分析软件和PLS_Toolbox工具箱对[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]进行如下步骤的处理和优化,最终建立水分水分定量分析模型。 考察两种常用数据增强算法:均值中心化(Mean Center)、标准化(Autoscaling);‚ 考察FD+SG、SD+SG平滑窗口宽度;ƒ 考察MSC、SNV、OSC预处理方式;④考察FiPLS、BiPLS、以及CARS方法选取特征波段;⑤采用PLS法建立桂皮醛定量校正模型,以校正集和验证集样品的RMSEC、RMSECV、[i]R[sup]2[/sup]c、[/i]RMSEP、[i]R[sup]2[/sup]p[/i]、LVs为指标,优化建模参数。⑥采用配对t检验法对预测值与测定值进行差异显著性检验,进一步评价模型准确性。均值中心化、标准化两种数据增强方式,均优于无处理方式,Mean Center较优,因此在下述处理中mean Center为基础处理方式。FD+S-G最佳平滑窗口宽度为3,SD+S-G最佳平滑窗口宽度为15,因此在接下来的数据处理中,均以最佳平滑窗口数进行。通过对不同预处理方式的考察,数据中可以看出最优处理方式为SNV+FD和FD。接下来以SNV+FD、FD分别为光谱预处理方式,进行特征波段选择。特征波段选择,采用FiPLS和CARS。预处理方式为FD,FiPLS-300即间隔数为300时,所选的波段区间建模模型RMSEP 最小RMSEC相对较小,[i]R[sub]c[/sub][/i][sup]2[/sup]、[i]R[sub]p[/sub][sup]2[/sup][/i]最大,结果最佳,且变量数最少。该方法对应光谱区间选择结果如图3-5所示,图形横坐标为波长变量 4000-10000 cm[sup]-1[/sup] 之间划分的3112个变量顺序,绿色区域对应 RMSECV 最小,即为所选变量区间6527.86-5951.25 cm[sup]-1[/sup],共包含300个变量,较全光谱缩减了2812个变量,改善模型结果的同时,降低90%的运算量。[align=center][img=,560,420]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261514457629_3089_3389662_3.jpg!w560x420.jpg[/img][/align][align=center]图 3-5FD,FORWARD iPLS-300 波段选择结果(实验记录Ⅱ-p108)[/align]以CARS法进行变量选择时对模型结果影响较大的两个参数为蒙特卡洛采样次数以及LVs,LVs考察2-10,蒙特卡洛采样次数考察10、25、50、100、200、500,以模型的RMSECV+RMSEP为评价参数。CARS前对图谱进行SNV+FD预处理考察结果见表3-3。[align=center][img=,587,410]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261515274590_6583_3389662_3.png!w587x410.jpg[/img][/align][align=center][/align]当LVs为7,采样次数为25时和LVs为8,采样次数为200时RMSECV+RMSEP处在较低水平。因此以这两个参数分别进行CARS波段选择,以FD为预处理方式,进行建模,模型评价见表3-5。[align=center][img=,558,137]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261516068540_2384_3389662_3.png!w558x137.jpg[/img][/align][align=center][img=,449,508]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261516196931_6560_3389662_3.png!w449x508.jpg[/img][/align][align=center][img=,431,291]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261517021361_7470_3389662_3.png!w431x291.jpg[/img][/align][align=center][img=,442,543]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261517366951_1045_3389662_3.png!w442x543.jpg[/img][/align][align=center][img=,447,230]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261519232531_9846_3389662_3.png!w447x230.jpg[/img][/align]最佳建模方式为FD+mean Center,FiPLS-300,模型参数为[i]R[sup]2[/sup]c[/i]=0.964,[i]R[sup]2[/sup]p[/i]=0.962,RMSEC=0.14419,RMSEP=0.13736,LVs为3。图3-10为水分预测值与实测值相关图。[align=center][img=,492,243]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261520051238_4887_3389662_3.png!w492x243.jpg[/img][/align]以PLS法建立的最佳模型计算得到的验证集样品的水分预测值和甲苯法测定的结果进行配对t检验,以评价模型的预测能力。表3-8为配对t检验的统计学结果,可见甲苯测定结果的平均值和NIRS得到的结果均值相同。在95%的置信限下,桂皮醛模型的P=0.560.05,说明近红外模型预测的结果和甲苯法的测定结果没有显著性差异,证实了NIRS用于桂枝药材水分测定的有效性。[align=center][img=,569,144]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261520481091_6921_3389662_3.png!w569x144.jpg[/img][/align][b](3)浸出物含量定量分析模型建立[/b]用Matlab化学计量学分析软件和PLS_Toolbox工具箱对[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]进行如下步骤的处理和优化,最终建立浸出物含量定量分析模型。比较两种常用数据增强算法:Mean Center、Autoscaling;考察FD+SG、SD+SG平滑窗口宽度;考察MSC、SNV预处理方式;考察FiPLS、BiPLS方法选取特征波段;采用PLS法建立浸出物定量校正模型,以校正集和验证集样品的RMSEC、RMSECV、[i]R[sup]2[/sup]c、[/i]RMSEP、[i]R[sup]2[/sup]p[/i]、LVs为指标,优化建模参数。采用配对t检验法对预测值与测定值进行差异显著性检验,进一步评价模型准确性。[align=center][/align][align=center][img=,566,144]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261521414114_9838_3389662_3.png!w566x144.jpg[/img][/align] 从表3-7知均值中心化(Mean Center)、标准化(Autoscaling)两种数据增强方式,均优于无处理方式,Autoscaling较优,因此在下述处理中Autoscaling为基础处理方式。表3-8为FD+SG、SD+SG平滑窗口宽度建模效果。由表3-8数据可知,FD+S-G最佳平滑窗口宽度为7,SD+S-G最佳平滑窗口宽度为15,因此在接下来的数据处理中,均以最佳平滑窗口数进行。以下表格中FD、SD均指FD+S-G(7)和SD+S-G(15)。[align=center][/align][align=center] [img=,567,417]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261522089231_2342_3389662_3.png!w567x417.jpg[/img][/align][align=center] [img=,542,460]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261522357719_3272_3389662_3.png!w542x460.jpg[/img][/align]通过对不同预处理方式的考察,在表3-9汇总的数据中可以看出最优处理方式为SNV+SD。以SNV+SD为光谱预处理方式,进行特征波段选择。特征波段选择,采用iPLS。[align=center][/align][align=center] [img=,546,644]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261523113364_1649_3389662_3.png!w546x644.jpg[/img][img=,544,160]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261523275961_9034_3389662_3.png!w544x160.jpg[/img][/align]从表3-10可以看出预处理方式为SNV+SD,BiPLS-250即间隔数为250时,所选的波段区间建模模型RMSEP 最小RMSEC相对较小,[i]R[sub]c[/sub][/i][sup]2[/sup]、[i]R[sub]p[/sub][sup]2[/sup][/i]最大,结果最佳。该方法对应光谱区间选择结果如图3-11所示,图形横坐标为波长变量 4000-10000 cm[sup]-1[/sup] 之间划分的3112个变量顺序,绿色区域对应 RMSECV 最小,即为所选变量区间9999.1-9518.91 cm[sup]-1[/sup]、8070.63-7108.33 cm[sup]-1[/sup]、5660.05-4697.75 cm[sup]-1[/sup]及4213.7-3999.64 cm[sup]-1[/sup],共包含1362个变量,较全光谱缩减了1750个变量,改善模型结果的同时,降低56%的运算量。[img=,242,182]http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif[/img][img=,538,231]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261524165981_3520_3389662_3.png!w538x231.jpg[/img]浸出物最佳建模方式为SNV+SD+Autoscaling,BiPLS-250,模型参数为[i]R[sup]2[/sup]c[/i]=0.967,[i]R[sup]2[/sup]p[/i]=0.900,RMSEC=0.22104,RMSEP=0.3763,LVs为3。图3-10为浸出物预测值与实测值相关图。以PLS法建立的最佳模型计算得到的验证集样品的浸出物预测值和药典法测定的结果进行配对t检验,以评价模型的预测能力。表3-11为配对t检验的统计学结果,可见药典法测定结果的平均值和NIRS得到的结果均值相同。在95%的置信限下,桂皮醛模型的P=0.240.05,说明近红外模型预测的结果和药典法的测定结果没有显著性差异,证实了NIRS用于桂枝药材浸出物测定的有效性。[align=center][img=,577,146]http://ng1.17img.cn/bbsfiles/images/2018/07/201807261524550178_7178_3389662_3.png!w577x146.jpg[/img][/align][b] 4总结[/b]通过收集市场上不同批次的桂枝样品,用常规方法测定桂皮醛、浸出物和水分的含量。桂皮醛、浸出物和水分的含量范围分别在0.543% ~1.83%、2.09% ~7.72 %和8.38 % ~11.09 %。药典规定桂皮醛、浸出物和水分的合格限为大于等于1.0%、大于等于6.0 %(作为参考)和不得过12 %。可见,市场上桂枝水分含量也基本稳定,而桂皮醛则存在不合格现象。不合格批次33批,占比44 %以上。说明市场上桂枝的品质存在很大的问题,这些与桂枝的产地、采收时间、加工方式不无关系,因此对于入库验收、对投料比例的把握就会提出更加严格的要求,光靠传统经验显然不足,常规方法又费时费力。开发快检方法尤为迫切。本实验成功运用 Antaris II傅立叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]以及相关化学计量学软件和方法建立了桂枝药材中桂皮醛、浸出物和水分的定量分析模型。基于Antaris II[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]的桂枝药材光谱经SNV+SD+Autoscaling,BiPLS-250组合处理,在9999.1-9518.91 cm[sup]-1[/sup]、8070.63-7108.33 cm[sup]-1[/sup]、5660.05-4697.75 cm[sup]-1[/sup]及4213.7-3999.64 cm[sup]-1[/sup]区间,所建 PLS模型最佳,桂皮醛水分最佳PLS模型参数为[i]R[sup]2[/sup]c[/i]=0.9855,[i]R[sup]2[/sup]p[/i]=0.9601,RMSEC=0.0427,RMSEP=0.0487,LVs为5;水分最佳PLS模型参数为[i]R[sup]2[/sup]c[/i]=0.964,[i]R[sup]2[/sup]p[/i]=0.962,RMSEC=0.14419,RMSEP=0.13736,LVs为3;浸出物最佳PLS模型参数为[i]R[sup]2[/sup]c[/i]=0.967,[i]R[sup]2[/sup]p[/i]=0.900,RMSEC=0.22104,RMSEP=0.3763,LVs为3。为桂枝药材的购买、筛选提供参考方法,保障投料稳定均一,从源头保障产品质量。
3,3-二甲氧基戊烷的理化性质,和供应厂商。急啊。
[align=right][b]SGLC-GC-003[/b][/align][b]摘要:[/b]本文建立了肉桂油中桂皮醛的检测方法。结果表明,采用色谱柱SH-5 (1.0um*0.53mm*30m)分析肉桂油中的桂皮醛,理论板数按桂皮醛峰计算为133586,满足《中国药典》要求。此方法可为肉桂油中的桂皮醛测定提供参考。[b]关键词:[/b]桂皮醛 SH-5[b]1. 实验部分1.1 实验仪器及耗材[/b]GC-FID[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-氢火焰离子化检测器;色谱柱:SH-5 (1.0um*0.53mm*30m;P/N 221-75710-30);SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05);[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34002-01);SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02);SHIMSEN Pipet PMII-100(P/N:380-00751-04);SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。[b]1.2 分析条件[/b]色谱柱:SH-5 (1.0um*0.53mm*30m)柱温:初始温度为100℃:,以每分钟5℃的速率升温至150℃,保持5分钟,再以每分钟5℃的速率升温至200℃,保持5分钟;载气:氮气进样口:200°C 分流比20:1检测器:220°C进样量:1 μL[b]2.结果及讨论2.1 色谱图[/b]按照上述色谱条件(1.2)进行采集,色谱图如下:[img=肉桂油中的桂皮醛]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-006_1.png[/img][b]3. 结论[/b]参考《中国药典》中色谱条件,并对其条件进行优化,最终建立了肉桂油中的桂皮醛的检测方法。结果表明,采用色谱柱SH-5 (1.0um*0.53mm*30m)分析肉桂油中的桂皮醛,理论板数按桂皮醛峰计算为133586,满足《中国药典》要求。此方法可为肉桂油中的桂皮醛测定提供参考。
气相测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
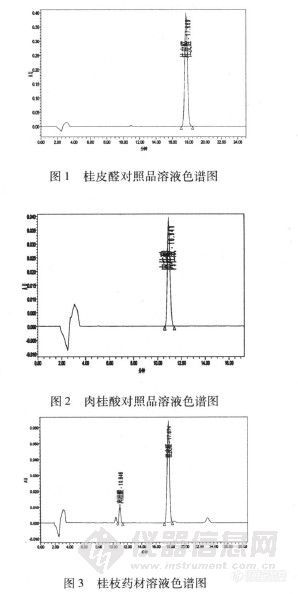
【作者】 王连芝; 蒋维谦;【机构】 黑龙江中医药大学中医药研究院;【摘要】 目的:建立HPLC法测定桂枝中桂皮醛和肉桂酸含量。方法:采用Diamonsil C18(250mm×4.6mm,5μm)色谱柱,以乙腈-0.1%磷酸溶液(38:62)为流动相,流速为1.0ml.min-1,检测波长为276nm和289nm双波长扫描。结果:样品中桂皮醛的平均回收率为99.48%,RSD为1.21%;肉桂酸的平均回收率为98.76%,RSD为1.29%;桂皮醛在0.01~0.03之间峰面积与浓度线性关系良好(r=0.9998);肉桂酸在0.002~0.01μg之间峰面积与浓度线性关系良好(r=0.9997)。结论:该实验方法简便,重现性好,回收率高,可作为同时测定桂枝中桂皮醛和肉桂酸含量的方法。 更多还原【关键词】 桂皮醛; 肉桂酸; 高效液相色谱法; 桂枝; 【基金】 黑龙江中医药大学科研基金项目(200745)http://ng1.17img.cn/bbsfiles/images/2012/08/201208071034_382135_2352694_3.jpg
[color=#444444]测定白油中环己基甲基二甲氧基硅烷的浓度,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]外标法,重复性很差,想请教一下问题在哪里?[/color][color=#444444]色谱条件简单如下:岛津GC-2014,无自动进样器[/color][color=#444444]柱温:100℃;进样口200℃;检测器300℃;[/color][color=#444444]程序升温:100℃(1min)--5℃/min升至150℃(0min)—25℃/min升至200℃(5min)[/color][color=#444444]色谱柱为非极性毛细管柱[/color][color=#444444]分流比:80:1[/color][color=#444444]样品采用正己烷稀释10倍后分析,质量浓度约15%,色谱分析重复性很差,做过此类分析的高手给些建议,谢谢![/color]
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
以 猪肉香肠为研究对象,研究桂皮、洋葱和茶多酚复配对阻断产品中N一亚硝胺生成的效果。实验结果表明:茶多酚对猪肉发酵香肠中亚硝胺残留量的影响最大,茶多酚、洋葱和桂皮三种物质复配能有效地抑制N一亚硝胺的生成,其最佳配比为(加人量以肉重计):茶多酚含量为0.029%,洋葱汁为3.5%, 6%桂皮浸体液含量为5.4%,此时产品中亚硝胺的含量6.5ug/kg
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]质谱有有一198.8001质谱峰,分析可能是是邻苯二甲酸二甲酯的去甲氧基后加Cl的峰(但准确数值是198.0083),请问是邻苯二甲酸二甲酯这个物质吗?
仪器走了10ppm 的,3,3二甲氧基联苯胺走的不好,啥情况呢?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2024/01/202401131427369784_1355_3971926_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2024/01/202401131427375863_7057_3971926_3.png[/img]
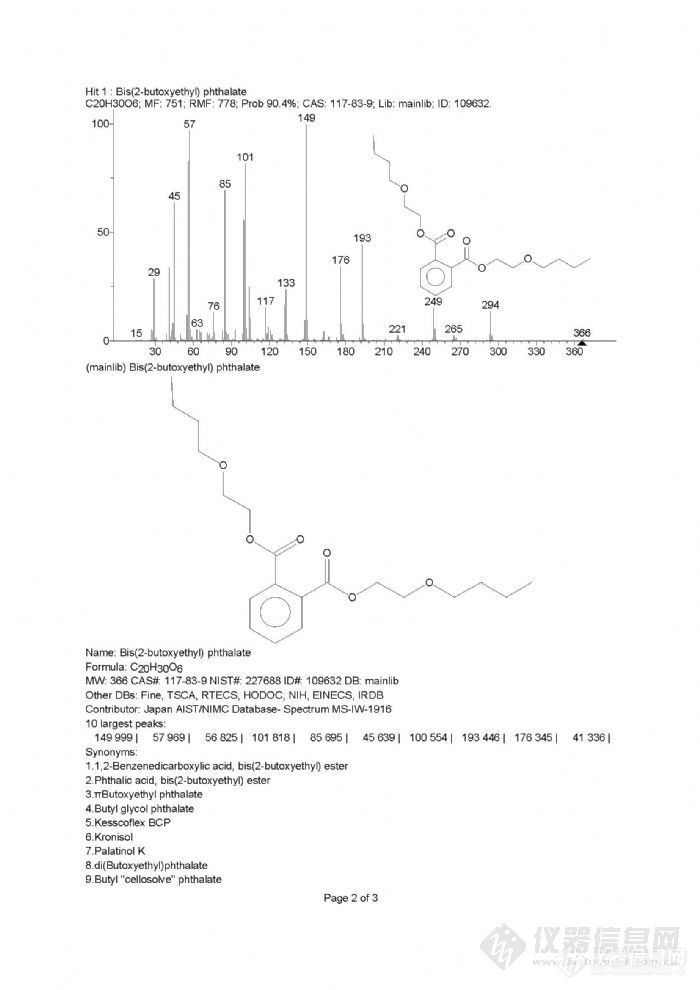
GB/T 21911-2008 《食品中邻苯二甲酸二甲酯的测定》中说明邻苯二甲酸二丁氧基乙基酯(DBEP)的特征离子为149(100) 223(14) 205(9) 278(3),但我进的标准品丰度与标准差很多。下图为我的DBEP标准品SIM图http://ng1.17img.cn/bbsfiles/images/2011/07/201107271350_307054_1644700_3.jpg再对DBEP标准品通过全扫描并与NIST库比对,几乎与谱库中DBEP质谱图完全符合下图为我进的标准品SCAN与NIST图http://ng1.17img.cn/bbsfiles/images/2011/07/201107271352_307056_1644700_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/07/201107271352_307058_1644700_3.jpg从上图可以看出根本没有205、278这两个的离子的,国标怎么会选呢?请教做过的、有经验的版友,谢谢!

【作者】 雷灼雨; 巴国际;【机构】 重庆市药品检验所; 重庆市药品检验所 重庆 400015; 重庆 400015;【摘要】 目的建立生桂口服液中桂皮醛的含量测定方法。方法采用反相高效液相色谱法,色谱柱为Diamonsil C18柱(150 mm×4.6 mm, 5μm),甲醇-水-冰醋酸(45:55:0.5)为流动相,流速为1.0 mL/min,测定波长为274 nm。结果方法的平均回收率为99.39%,RSD= 0.27%,桂皮醛的线性范围是25.2~201.4μg/mL。结论所建立的方法准确、可靠,能满足该产品的质量控制要求。 更多还原【关键词】 生桂口服液; 桂皮醛; 反相高效液相色谱法; http://ng1.17img.cn/bbsfiles/images/2012/08/201208271555_386456_2352694_3.jpg
[em0808] [em0808] 各位求助一下关于2.5-二甲基-2.5-双(叔丁基过氧基)己烷的测定,性质,反正关于2.5-二甲基-2.5-双(叔丁基过氧基)己烷的任何资料都可以了,在网上都查过了,找到一篇,我先发上来大家看看.有的话麻烦大家分享一下了[~82374~]
请教:乙烯基甲醚、丙烯醛、二甲氧基二氢吡喃、戊二醛这几种物质可否有[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测?用什么样的柱子和检测条件?
桂皮油和肉桂油有什么区别啊
请问1,2-丙二醇单甲醚和甲氧基丙酮混合物中甲氧基丙酮的含量如何分析?谢谢!
(符合《中华人民共和国药典》 2010年版一部 P127 肉桂) 样品制备 制备方法:含量测定:桂皮醛对照品溶液的制备:取桂皮醛对照品适量,用甲醇溶解,配成浓度为11.66 μg / mL的溶液。 分析条件 色谱柱:Diamonsil C18(2),150×4.6 mm,5 μm (Cat#:99601)流动相:乙腈:水=35:65流速:1.0 mL/min柱温:30 ℃检测器:UV 290 nm进样量:10 μLhttp://www.dikma.com.cn/Public/Uploads/images/11(44).JPG 峰号保留时间min峰面积μV*s峰高μV理论塔板数NUSP拖尾因子分离度
请问,1,4-二甲氧基苯C8H10O2在254nm处,是不是没有吸收峰啊?谢谢了
第一步:甲氧基聚乙二醇的合成聚乙二醇在无水二氯甲烷中与金属钠作用生成聚乙二醇钠, 然后与碘甲烷反应即得。一甲氧基聚乙二醉、双端都反应的二一甲氧基聚乙止醇和未反应的聚乙二醇的反应混合物硅胶柱层析色潜提纯可以得到纯净的甲氧基聚乙二醇第二步:甲氧基聚乙二醇丁二酸单醋的合成将甲氧基聚乙二醇(Me-PEG-2000)、丁二酸酐和催化剂加入盛有二氯甲烷的圆底烧瓶中, 磁力搅拌使固体完全溶解后, 室温搅拌反应过夜。反应液分别用盐酸水溶液、氢氧化钠水溶液和甲醇水溶液依次洗涤。有机相经无水MgSO4干燥, 过滤除去干燥剂, 减压蒸除有机溶剂, 残留物以石油醚结晶, 收率90%。第三步:甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺的合成甲氧基聚乙二醇丁二酸单酷先经N一羟基丁二酰亚胺(NHS)活化, 然后缓慢滴加人到二硬脂酰磷脂酰乙醇胺(DSPE)的三氯甲烷中, 加料完毕后继续反应4h, 蒸除溶剂, 浓缩液在乙醚中结晶,硅胶柱层析色谱提纯可以得到自色粉末状固体的。甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺。来源:中国标准物质网
RT76.654+76.696是对甲氧基肉桂酸辛酯吗? CAS:5466-77-3 我这里匹配出来这个原料。但这个是防晒剂 ,怎么添加量这么大?
我们是做化妆品的企业,因为产品中有添加辛二醇、辛氧基甘油作为防腐剂,现需要检测其含量。但我们用甲醇加样品溶解后用HPLC测试却没有跑出任何峰出来,想请问一下各位高手:1.这二类物质是否可以用HPLC来检测? 2.是否我们的样品处理方法有问题? 3.对于不出峰的样品,大家是否有其他的一些样品处理方法或测试条件等各方面的建议?? 急!急!!急!!!急!!!!急!!!!![em0705]
1 试剂和对照品除特殊说明,所用试剂均为色谱纯。1.1 试剂:甲酸,甲醇,氯化钠(分析纯),乙腈1.2 对照品:邻苯二甲酸酯类混合对照(供含量测定用)、DNP(供含量测定用)2 仪器2.1 液相质谱联用仪(API3200)2.2 分析天平2.3 超声震荡器2.4 涡旋仪2.5 离心机2.6 氮吹仪3 分析方法3.1 色谱参考条件1-适用于DNP3.1.1 色谱柱:以十八烷基硅烷健合硅胶为填充剂(柱长5cm,内径4.6mm,粒径2.7μm )或同等性能的色谱柱3.1.2 柱温:40℃3.2 色谱参考条件2-适用于剩余的22种塑化剂3.2.1 色谱柱:以十八烷基硅烷健合硅胶为填充剂(柱长5cm,内径4.6mm,粒径2.7μm )或同等性能的色谱柱与以十八烷基硅烷健合硅胶为填充剂(柱长15cm,内径4.6mm,粒径5μm )或同等性能的色谱柱两根串联3.2.2 柱温:40℃3.2.3 流动相:时间(分钟)流速(ml/min)0.1%甲酸水溶液(%)甲醇(%)0.000.820802.000.820805.000.820808.000.829813.000.829813.101.229814.001.229814.011.329818.001.329818.100.8208023.000.820803.2.4 质谱条件:ESI+ 模式化合物 Q1Q3Time(msec)IDDPEPCECXP邻苯二甲酸酯二壬酯419.335149.100300DNP 166.06.0021.004.00 419.335275.200300DNP 260.45.6215.356.25邻苯二甲酸酯二甲酯195.116320DMP 11510163 195.113520DMP 21510353邻苯二甲酸二乙酯223.1149.120DEP 12010273 223.1121.120DEP 22010443邻苯二甲酸二丙酯251.214920DPrP12310173 251.2191.120DPrP22310173邻苯二甲酸二异丙酯251.214920DIPrP12310173 251.2121.120DIPrP22310173邻苯二甲酸二烯丙酯247.1189.120DAP 12010123 247.1149.120DAP 22010263邻苯二甲酸二异丁酯279.214920DIBP 12310223 279.2205.120DIBP 22310123邻苯二甲酸二丁酯279.214920DBP 12310223 279.2205.120DBP 22310123邻苯二甲酸二(2-甲氧基)乙酯283.2207.120DMEP 12810113 283.259.120DMEP 22810303邻苯二甲酸二异戊酯307.27120DIPP13010253 307.2149.120DIPP23010213邻苯二甲酸二(4-甲基-2-戊基)酯335.314920BMPP 13010273 335.3167.120BMPP 23010183邻苯二甲酸二(2-乙氧基)乙酯311.2221.120DEEP 13010123 311.273.120DEEP 23010213邻苯二甲酸二戊酸307.2149.120DPP13010213 307.2219.120DPP23010123邻苯二甲酸丁基苄基酯313.291.120BBP 13010403 313.2149.120BBP 23010213邻苯二甲酸二己酯335.314920DHXP 13010273 335.3233.120DHXP 23010123邻苯二甲酸二(2-丁氧基)酯367.3101.120DBEP 13510183 367.3249.120DBEP 23510123邻苯二甲酸二环己酯331.2149.120DCHP 13010363 331.2167.120DCHP 23010213邻苯二甲酸二庚酯363.214920DHP 13510223 363.2247.120DHP 23510133邻苯二甲酸二(2-乙基)己酯391.314920DEHP 13510253 391.3167.120DEHP 23510203邻苯二甲酸二苯酯319.122520D_P 13010163 319.17720D_P 23010543邻苯二甲酸二正辛酯391.314920DNOP 13510253 391.3261.220DNOP 23510133邻苯二甲酸二异壬酯419.314920DINP 13810543 419.3127.220DINP 23810183邻苯二甲酸二异癸酯447.214120DIDP 14510153 447.2289.220DIDP 24510153CURCADISTEMGS1GS2256550065065653.3 溶液制备3.3.1对照品溶液制备:3.3.1.1邻苯二甲酸酯二壬酯(DNP)对照液:精密称取邻苯二甲酸酯二壬酯(DNP)对照品5mg,置于500ml容量瓶中,加入适量甲醇超声溶解,定容。精密量取1ml置于100ml容量瓶中,加入甲醇定容至刻度,摇匀,过0.45μm的滤膜,即得。3.3.1.2邻苯二甲酸酯类混合对照液:精密量取邻苯二甲酸酯类混合对照品1ml,置于100ml容量瓶中,加入甲醇定容至刻度,摇匀,过0.45μm的滤膜,即得。3.3.2供试品溶液制备:3.3.2.1不含油脂固体或半固体试样 取固体试样20粒(片)或10g(若试样为胶囊,应取内容物),研细,混合均匀。精密称取试样1.0g,置于50ml具塞比色管中,精密量取5ml乙腈,超声(功率500W,频率50kHz)处理至提取完全(约5min),取上清液,经0.45μm的滤膜过滤,即得。3.3.2.2 含油脂试样 精密称取混合均匀试样1.0g,置于25ml具塞磨口玻璃试管中,加入5ml乙腈,涡旋混合2min, 超声(功率500W,频率50kHz)处理至提取完全(约10min),离心,收集上清液。重复提取一次,合并上清液。将提取液于40℃氮吹至干,精密量取1ml乙腈,超声(功率500W,频率50kHz)处理至提取完全(约1min),放入-18℃的冰箱冷藏2h,离心,取上清液,经0.45μm的滤膜过滤,即得。3.4 测定精密吸取对照品溶液进样量:2μL,4μL,6μL,8μL,10μL与供试品溶液进样量:10μl,注入液相质谱联用仪,建立标准曲线方程。供试品色谱中应呈现与对照品色谱峰保留时间相同的色谱峰定量。3.5 结果计算X=V×C×100/M式中: X—试样中邻苯二甲酸酯类的含量,mg/100g;C—试样溶液中邻苯二甲酸酯类的浓度,mg/mL; M—试样的质量,g; V—试样稀释的体积,mL。
求助:化妆品中的二甲基硅氧烷和混居二甲基硅氧烷如何提取啊?
各位大虾好,小第有一疑问盼大家给点意见。我做美国药典中的盐酸米托蒽醌中的残留溶剂时,按照厂家给的资料要检测其中的2-甲氧基乙醇,厂家给的方法为直接进样(水做溶剂)。但它的样品浓度为200mg/ml,按照盐酸米托蒽醌的溶解度看,样品是绝对不能完全溶解的,样品溶解后成米糊状,离心处理也处理不到上清液,进样发现样品出峰非常的难看,且不见残留溶剂峰。我们打算自己开发方法用顶空做,但美国药典467中建议2-甲氧基乙醇用直接进样法,各位大虾你有过内似的经历么,2-甲氧基乙醇用顶空法检测后,美国FDA能批准么?
各位大佬,桂皮醛对照品液体0.5ml(药典要求每1ml含10ug)这个要怎么换算去取呢?直接称重2mg的和直接吸2ul的,但是浓度都是0.01,跑出来峰面积相差好大,后者吸的2ul和样的峰面积都是7位数,前者称重的mg跑出来就只有6位数,算出来的结果就也不一样了,2ul的含量是结果2.7,与药典的1.5比较符合,后者称的mg结果就是10.5了 这个结果相差好大的
徐建强(南京气象学院环境科学系,南京 210044)分光光度法是获得物质光吸收特性及定性、定量分析的重要手段,在冶金、地质、生物、医学、农业、环境监测、食品卫生等部门得到极其广泛的应用。分光光度法的发展不仅依赖于电子学、激光和计算机技术的发展和应用,而且还依赖于高灵敏度、高选择性有机试剂的合成和应用。喹啉类试剂作为光度分析的有机试剂,可以分为两类,一类是喹啉及其衍生物,如8-羟喹啉、8-巯基喹啉、8-氨基喹啉等,这类试剂可用作金属离子光度分析的显色剂,具有一定的灵敏度和选择性 另一类是喹啉偶氮化合物,喹啉偶氮化合物作为一大类显色剂已有多种试剂被合成和研究。其中8-氨基喹啉的8位偶氮衍生物前苏联学者研究得较早 我国的李亚文等也进行了系统深入的研究,近年来不断有一系列新的衍生物合成,这些化合物因其特有的灵敏度和选择性而备受化学工作者的关注。自从1955年程广禄等[3]首次提出PAN(即1-(2-吡啶偶氮)-2-萘酚)作为分析试剂后,越来越多的偶氮试剂相继被化学工作者合成并且用于光度分析研究。实践证明,偶氮化合物(azo-compounds)具有性质稳定、显色反应灵敏度高、选择性好、对比度大等优点,仍然是目前应用最广泛的一类显色剂。本文选用6-甲氧基-8-氨基喹啉作为原料,首先对其进行重氮化,得到重氮盐,然后与间苯二酚进行偶联反应,合成了新有机试\剂:4-(6-甲氧基-8-喹啉偶氮)-间苯二酚(简称MQAR)。对其进行了结构分析和鉴定。实验研究表明,MQAR能与Co、Cu、Fe、Ni等金属离子发生灵敏的显色反应,该试剂可用于试样中微量金属离子的测定(另文介绍),方法简便、快速、准确可靠,是一种比较理想的新有机试剂。1 合成方法1.1 试剂和仪器设备6-甲氧基-8-氨基喹啉(由工业品提纯所得),亚硝酸钠(AR),间苯二酚(AR),N,N-二甲基甲酰胺(AR),浓硫酸(AR),甲酸(AR)。中量有机化学制备仪、真空干燥箱、差热分析仪、Perkin-Elmer元素分析仪、IR-408型红外分光光度计、756MC型紫外-可见分光光度计、BRUKERARX300M核磁共振谱仪。1.2 合 成1.2.1 合成线路MQAR的合成线路如图1所示。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221937_18814_1634962_3.gif[/img]1.2.2 合成步骤(1)重氮化 在250mL三口烧瓶中,加入8.7g6-甲氧基-8-氨基喹啉,10mL甲酸,同时加入由15mL浓硫酸和10mL水配制而成的溶液,搅拌溶解,置于冰浴中冷却。在0~5℃时边搅拌边滴加3.5g亚硝酸钠与10mL水配制而成的溶液,控制在1h内加完,使反应充分完全,最后得到深红色的重氮盐溶液,用于下一步的反应。(2)偶联 取5.5g间苯二酚溶于75mL无水乙醇中,置于冰浴中冷却。在0~5℃时,边搅拌边加入上述重氮盐溶液,控制重氮盐溶液于1h内加完,继续搅拌2h,静置过夜得砖红色沉淀,抽滤,依次用水、无水乙醇洗涤,抽干。干燥后得棕黄色粗品。(3)精制 将偶联得到的粗品用N,N-二甲基甲酰胺重结晶两次,于真空干燥箱干燥,得到深红色的MQAR纯品。经测定产品熔点为194℃。2 结构分析2.1 薄层色谱分析(1)试剂和仪器设备 展开剂正丁醇∶无水乙醇∶2molL-1氨水=3∶1∶1 支持剂硅胶HF254+0.3%CMC 8×10cm2薄层板(自制) 层析缸。(2)结果和讨论 在不同极性的溶剂体系中进行展开,在薄层色谱板上只发现一个斑点,在紫外灯光下未发现其他斑点,(1)中所列展开剂效果最好,比移值Rf=0.66(图2)。结果表明,产品中只含有一种物质。2.2 元素分析用Perkin-Elmer元素分析仪对产品进行了元素分析,产品MQAR元素分析结果与理论计算值基本一致。2.3 紫外-可见吸收光谱分析用756MC型紫外-可见分光光度计测得2×10-5molL-1MQAR的10%DMF水溶液(pH=8.3时)的紫外-可见吸收光谱(图3)。Kmax=450nm,E=2.98×104Lmol-1cm-1。结果表明,产品分子是一个大的共轭体系。2.4 红外吸收光谱分析用IR-408型红外分光光度计对产品MQAR进行了红外吸收光谱分析(KBr压片法),产品的红外光谱解析结果见表1。解析结果表明,产品分子中含有酚羟基、芳环、偶氮基等官能团,还具有1,2,4-三取代苯结构。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221954_18815_1634962_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18816_1634962_3.gif[/img]2.5 核磁共振谱分析用BRUKERARX300M核磁共振谱仪(DMSO-d6溶剂,TMS内标)对产品进行核磁共振谱分析,得到化学位移、峰面积等(表2)。解析结果表明,产品分子中除了羟基质子以外,还含有甲氧基质子以及苯环和杂环质子。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18817_1634962_3.gif[/img]2.6 产品的结构根据实验条件以及结构鉴定和分析,可以确定产品4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的分子结构为[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221956_18818_1634962_3.gif[/img]3 结 论研究了新有机试剂4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的合成方法、实验条件和精制方法,通过薄层色谱法、元素分析、紫外-可见吸收光谱法、红外吸收光谱法和核磁共振谱法等分析手段,对合成的产品进行了分析和结构鉴定,实验结果确证合成了4-(6-甲氧基-8-喹啉偶氮)-间苯二酚纯品。







 我要推广仪器
我要推广仪器
 下载APP
下载APP