[b]1 方法依据[/b]本方法依据GB 5009.188-2003《蔬菜、水果中甲基托布津、多菌灵的测定》。[b]2 方法原理[/b]用甲醇自试样中提取出甲基托布津,在pH 1~2时,用二氯甲烷提取,甲基托布津经闭环反应转变为多菌灵,提纯后,用紫外分光光度法进行定量测定。多菌灵经提取后可直接测定吸光度而进行定量。[b]3 试剂[/b]3.1 甲醇3.2 二氯甲烷3.3 三氯甲烷3.4 石油醚:沸程30℃~60℃3.5 乙酸-乙酸铜溶液:称取2g乙酸铜,加100ml冰乙酸,稍加热溶解,用水稀释至200ml3.6 盐酸(1+11):量取盐酸90ml,用水稀释至1000ml3.7 氢氧化钠溶液(80g/L):称取8g氢氧化钠,加水溶解并稀释至100ml3.8 氢氧化铵溶液(1+7):量取氨水10ml,加水稀释至80ml3.9 氯化钠溶液(100g/L)3.10 甲基托布津标准溶液:准确称取50.0mg甲基托布津,置于烧杯中,用三氯甲烷溶解并转移至50ml容量瓶中,稀释至刻度。此溶液每毫升相当于1.0mg甲基托布津。3.11 甲基托布津标准使用液:吸取10.0ml甲基托布津标准溶液置于100ml容量瓶中,加三氯甲烷稀释至刻度,此溶液每毫升相当于100.0ug甲基托布津。3.12 多菌灵标准溶液:准确称取50.0mg多菌灵置于烧杯中,用盐酸(1+11)溶解移入50ml容量瓶中,并稀释至刻度,此溶液每毫升相当于1.0mg多菌灵。3.13 多菌灵标准使用液:吸取10.0ml多菌灵标准溶液,置于100ml容量瓶中,加盐酸(1+11)稀释至刻度。此溶液每毫升相当于100.0ug多菌灵。[b]4仪器[/b]4.1 空气冷凝管4.2 紫外分光光度计[b]5分析步骤[/b]5.1 标准曲线的制备5.1.1甲基托布津标准曲线:吸取0、0.10、0.30、0.50ml甲基托布津标准使用液(相当于0、10、30、50ug甲基托布津),分别置于30ml圆底离心管中,挥干溶剂后,各加10ml乙酸-乙酸铜溶液及2粒玻璃珠,接上空气冷凝管,小火缓缓煮沸0.5h,取下,用20ml盐酸(1+11)从冷凝管顶端洗涤冷凝管和圆底离心管,并移入125ml分液漏斗中,用二氯甲烷提取二次,每次10ml,弃去二氯甲烷层,酸溶液中加25ml氢氧化钠溶液(80g/L)至pH6.0~6.5(pH试纸试),用二氯甲烷提取二次,每次20ml,合并二氯甲烷提取液,用10ml水洗涤一次,静置分层后,将二氯甲烷层分入另一个干的分液漏斗中,准确加入10ml盐酸(1+11),振摇5min,静置分层后,盐酸提取液用1cm石英比色杯,以盐酸(1+11)调节分光光度计零点,测读250nm~300nm的吸光度,以波长为横坐标,吸光度为纵坐标,绘制吸收图谱。将图谱上260nm和290nm吸光度读数点连成直线,设直线上282nm的吸光度A,两者之差为ΔA(ΔA=A-A’,为校正吸光度)。再以校正吸光度为纵坐标,甲基托布津的含量为横坐标,绘制各甲基托布津标准点ΔA值的标准曲线。5.1.2多菌灵标准曲线:吸取0、0.10、0.30、0.50ml多菌灵标准使用液(相当于0、10、30、50ug多菌灵),置于盛有20ml盐酸(1+11)的分液漏斗中,各用二氯甲烷提取二次,每次10ml,弃去二氯甲烷层,水溶液用氢氧化铵(1+7)中和到pH为6.0~6.5(pH试纸试),用二氯甲烷提取二次,每次20ml,提取液用10ml水洗涤一次,以下按甲基托布津自“将二氯甲烷层分入另一个干的分液漏斗中,准确加入10ml盐酸(1+11)”起依法操作,并绘制吸收图谱,计算出ΔA值后,绘制多菌灵的标准曲线。5.2 试样的提取和分离称取50.0g切碎、混匀的试样,加50ml甲醇振摇0.5h,用布什漏斗抽滤,容器和滤器用甲醇洗涤二次,每次15ml~20ml,抽干后,滤液移入烧杯中,抽滤瓶用约10ml水洗涤,洗液并入滤液内,在水浴上用空气流吹去部分甲醇后,移入分液漏斗中,加30ml氯化钠溶液(100g/L),用石油醚振摇提取二次,每次25ml,弃去石油醚,加盐酸酸化至pH为1~2(用pH试纸试),用二氯甲烷提取二次,每次25ml,合并二氯甲烷提取液,用25ml水洗涤一次分出二氯甲烷层留作甲基托布津测定用。水洗涤液合并入水层,留作多菌灵测定用。5.3 甲基托布津的测定二氯甲烷提取液自然挥干后,用10ml乙酸-乙酸铜溶液分次溶解残渣,并转入30ml圆底离心管中,加2粒玻璃珠,以下按5.1.1自“接上空气冷凝管”起依法操作,计算出试样的ΔA值,再与甲基托布津的标准曲线比较,计算试样中的含量。5.4 多菌灵的测定 取5.2中留作多菌灵测定的水溶液,再氢氧化铵(1+7)中和至pH6.0~6.5,然后按5.1.2多菌灵标准曲线“用二氯甲烷提取二次”起依法操作,计算出试样中ΔA值,再与多菌灵标准曲线比较,计算试样中的含量。[b]6结果计算[/b]试样中甲基托布津、多菌灵含量按下式计算: X=m′×1000/m×1000式中: X —试样中甲基托布津、多菌灵含量,单位为毫克每千克(mg/kg)m′[i]—[/i]样品中甲基托布津、多菌灵含量,单位为微克(ug);m —试样的质量,单位为克(g);计算结果表示到两位有效数字。[b]7试验结果报告[/b]7.1校准曲线及线性范围: [table][tr][td=1,3] [align=center]工作[/align] [align=center]曲线[/align] [/td][td] [align=center]顺序号[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]4[/align] [/td][/tr][tr][td] [align=center]质量( ug )[/align] [/td][td] [align=center]0.00[/align] [/td][td] [align=center]10.00[/align] [/td][td] [align=center]30.00[/align] [/td][td] [align=center]50.00[/align] [/td][/tr][tr][td] [align=center]吸光值(A)[/align] [/td][td] [align=center]0.000[/align] [/td][td] [align=center]0.035[/align] [/td][td] [align=center]0.093[/align] [/td][td] [align=center]0.163[/align] [/td][/tr][tr][td=2,1] [align=center]回归方程[/align] [/td][td=4,1] [align=center]y = 0.0032x +0.0004[/align] [/td][/tr][tr][td=2,1] [align=center]相关系数[/align] [/td][td=4,1] [align=center]0.9984[/align] [/td][/tr][/table]7.2 方法的精密度称取约50.0g样品,按照步骤5处理,平行制备9份样品,计算的含量并求出相对标准偏差,结果见下表所示。 [table=620][tr][td=2,1] [align=center]顺序号[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]4[/align] [/td][td] [align=center]5[/align] [/td][td] [align=center]6[/align] [/td][td] [align=center]7[/align] [/td][td] [align=center]8[/align] [/td][td] [align=center]9[/align] [/td][/tr][tr][td=1,4] [align=center]1[/align] [/td][td] [align=center]取样量(g)[/align] [/td][td] [align=center]50.0176[/align] [/td][td] [align=center]50.0229[/align] [/td][td] [align=center]50.0010[/align] [/td][td] [align=center]50.0098[/align] [/td][td] [align=center]50.0076[/align] [/td][td] [align=center]50.0089[/align] [/td][td] [align=center]50.0006[/align] [/td][td] [align=center]50.0097[/align] [/td][td] [align=center]50.0011[/align] [/td][/tr][tr][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.043[/align] [/td][td] [align=center]0.042[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.04[/align] [/td][td] [align=center]0.042[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.040[/align] [/td][/tr][tr][td] [align=center]多菌灵含量(ug)[/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]14.69 [/align] [/td][td] [align=right]14.38 [/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]13.75 [/align] [/td][td] [align=right]14.38 [/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]13.75 [/align] [/td][/tr][tr][td] [align=center]浓度 mg/kg[/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.27 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.25 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.25 [/align] [/td][/tr][tr][td] [align=center]平均值[/align] [/td][td=10,1] [align=center]0.26 (S=0.0061)[/align] [/td][/tr][tr][td]相对标准偏差 [align=center]RSD(%)[/align] [/td][td=10,1] [align=center]2.34[/align] [/td][/tr][tr][td=2,1] [align=center]备注[/align] [/td][td=9,1] [align=center]结论:九个平行样品的相对标准偏差为2.34%,小于5%,符合要求。[/align] [/td][/tr][/table]7.3方法的中间精密度随机选取2名实验人员,按照步骤5测定砷的含量,每个实验者平行测定3份样品,求出两者实验结果的相对标准偏差,结果见下表所示。[align=center]表2 中间精密度实验结果[/align] [table][tr][td] [align=center]实验者[/align] [/td][td] [align=center]多菌灵含量(mg/kg)[/align] [/td][td] [align=center]平均结果(mg/kg)[/align] [/td][td] [align=center]相对标准偏差(%)[/align] [/td][/tr][tr][td=1,3] [align=center] [/align] [align=center]甲[/align] [/td][td] [align=center]0.26[/align] [/td][td=1,3] [align=center]0.26[/align] [/td][td=1,6] [align=center]2.99[/align] [/td][/tr][tr][td] [align=center]0.25[/align] [/td][/tr][tr][td] [align=center]0.26[/align] [/td][/tr][tr][td=1,3] [align=center] [/align] [align=center]乙[/align] [/td][td] [align=center]0.24[/align] [/td][td=1,3] [align=center]0.25[/align] [/td][/tr][tr][td] [align=center]0.25[/align] [/td][/tr][tr][td] [align=center]0.25[/align] [/td][/tr][tr][td=4,1] 接受标准:从精密度测试所得的6组数据中间精密度测试的相对标准偏差应小于5.0%。[/td][/tr][/table]7.4方法的准确度分别向试样中加入一定量的多菌灵,使其砷的加标浓度为10ug/L的样品2ml、分别制备3份样品,按照步骤5处理,同时测定未加标样品硫脲含量,分别计算回收率、结果见下表所示。 [table][tr][td=3,1] [align=center]样品[/align] [/td][td=3,1] [align=center]标准品[/align] [/td][td=3,1] [align=center]样品加标测量值[/align] [/td][td=1,2] [align=center]回收率(%)[/align] [/td][td=1,2] [align=center]平均[/align] [align=center]回收率(%)[/align] [/td][/tr][tr][td] [align=center]浓度(mg/kg)[/align] [align=center] [/align] [/td][td] [align=center]取样量(g)[/align] [/td][td] [align=center]多菌灵含量[/align] [align=center](ug)[/align] [align=center]M[sub]1[/sub][/align] [/td][td] [align=center]浓度(ug/ml)[/align] [/td][td] [align=center]取样量(ml)[/align] [/td][td] [align=center]M[sub]2[/sub][/align] [align=center](ug)[/align] [/td][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]浓度[/align] [align=center](mg/kg)[/align] [/td][td] [align=center]质量[/align] [align=center]M[/align] [/td][/tr][tr][td] [align=right]0.252 [/align] [/td][td] [align=center]50.0305[/align] [/td][td] [align=center]12.6[/align] [/td][td] [align=center]10[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]20[/align] [/td][td] [align=center]0.331[/align] [/td][td] [align=center]0.63[/align] [/td][td] [align=right]32.81 [/align] [/td][td] [align=center]101.0[/align] [/td][td=1,3] [align=center]102.1[/align] [/td][/tr][tr][td] [align=right]0.248[/align] [/td][td] [align=center]50.0725[/align] [/td][td] [align=center]12.4[/align] [/td][td] [align=center]10[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]20[/align] [/td][td] [align=center]0.340[/align] [/td][td] [align=center]0.64[/align] [/td][td] [align=right]33.13 [/align] [/td][td] [align=center]103.6[/align] [/td][/tr][tr][td] [align=right]0.256 [/align] [/td][td] [align=center]50.0024[/align] [/td][td] [align=center]12.8[/align] [/td][td] [align=center]10[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]20[/align] [/td][td] [align=center]0.322[/align] [/td][td] [align=center]0.64[/align] [/td][td] [align=right]33.13 [/align] [/td][td] [align=center]101.6[/align] [/td][/tr][tr][td=3,1] [align=center]备注[/align] [/td][td=8,1] [align=center]△%=(M- M[sub]1[/sub])*100/ M[sub]2[/sub][/align] [/td][/tr][/table]7.5 检出限DL=0.02mg/kg取20次平行测定空白样的结果,按IUPAC规定DL=KSb/50a其中K=3Sb:空白多次测得信号的标准偏差:0.0008a: 校准曲线的斜率:0.0032 [table=595][tr][td] [align=center]顺序号[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]4[/align] [/td][td] [align=center]5[/align] [/td][td] [align=center]6[/align] [/td][td] [align=center]7[/align] [/td][td] [align=center]8[/align] [/td][td] [align=center]9[/align] [/td][td] [align=center]10[/align] [/td][/tr][tr][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][/tr][tr][td] [align=center]顺序号[/align] [/td][td] [align=center]11[/align] [/td][td] [align=center]12[/align] [/td][td] [align=center]13[/align] [/td][td] [align=center]14[/align] [/td][td] [align=center]15[/align] [/td][td] [align=center]16[/align] [/td][td] [align=center]17[/align] [/td][td] [align=center]18[/align] [/td][td] [align=center]19[/align] [/td][td] [align=center]20[/align] [/td][/tr][tr][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.008[/align] [/td][/tr][tr][td] [align=center]Sb[/align] [/td][td=10,1] [align=center]0.0008[/align] [/td][/tr][tr][td] [align=center]检出限(μg/g )[/align] [/td][td=10,1] [align=center]0.02[/align] [/td][/tr][tr][td] [align=center]公式[/align] [/td][td=10,1] [align=center]DL=f*K*Sb/50a ( K=3, Sb:空白信号的标准偏差,a:曲线斜率)[/align] [/td][/tr][tr][td] [align=center]备注[/align] [/td][td=10,1] [align=center]DL=K*Sb/50a [/align] [/td][/tr][/table][b] [/b]8 是否对方法偏离?是□否■[b]9 结论[/b]我公司对[u]蔬菜、水果中甲基托布津、多菌灵的测定 [/u]的检测符合G[u]B 5009.188-2003[/u]要求。
甲基硫菌灵(甲基托布津),在现行有效的国内的检测标准中,有[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]的标准吗?有知道的老师说一下的。
求助各位老师,有没使用过 氯化甲基汞 汞的同位素 标准品,或是做过水产品中甲基汞检测的,麻烦告知一下,谢谢?
蔬菜、水果及其制品中吡虫啉、多菌灵、甲基硫菌灵、霜霉威、灭多威、霜脲氰残留量的检测方法-超高效液相色谱-质谱/质谱法 唐玉萍1 范围本非标方法规定了蔬菜、水果及其制品中吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰和灭多威残留量的超高效液相色谱-质谱/质谱检测方法。本非标方法适用于蔬菜、水果及其制品中吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰和灭多威残留量的检测,该方法在番茄、番茄酱、梨、脱水洋葱等样品中经过验证。2 规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法3 原理样品用乙酸-甲醇-水溶液提取,C18色谱柱进行分离,用超高效液相色谱-质谱/质谱检测,内标或外标法定量。4 试剂及材料除另有规定外,所用试剂均为分析纯,水为GB/T 6682 规定的一级水。4.1 甲醇:色谱纯。4.2 乙酸铵。4.3 10mmol/L乙酸铵:准确称取1.926g乙酸铵,定容至500mL容量瓶中,配制成50mmol/L乙酸铵。用溶剂过滤装置过0.2µm水相滤膜,0~4℃保存,有效期15天。临用时用水稀释成10mmol/L。4.4 冰乙酸。4.5 甲醇-水溶液(30+70,V/V)。4.6 提取液Ⅰ:乙酸-甲醇-水溶液(0.1+50+50,V/V/V)。4.7 提取液Ⅱ:乙酸-甲醇-水溶液(0.1+80+20,V/V/V)。4.8 吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰、灭多威和D4-吡虫啉标准品:均为德国Dr.公司,纯度≥98.0%。4.9 6种农残标准储备液:分别准确称取吡虫啉、多菌灵、甲基硫菌灵、霜霉威、霜脲氰、灭多威标准品10mg~15mg(精确到0.1mg)于50mL容量瓶中,用甲醇溶解并定容至刻度,配制成浓度约为200~300µg/mL的标准储备液,于-18℃避光保存,有效期18个月。4.10 中间浓度混合标准溶液:根据需要,取适量6种农残标准储备液,用甲醇-水溶液(4.5)稀释配制成2µg/mL的混合标准中间液,0~4℃保存,有效期6个月。4.11 内标标准储备液:准确称取D4-吡虫啉标准品约10mg(精确到0.1mg)于50mL容量瓶中,用甲醇溶解并定容至刻度,配制成浓度约为200µg/mL的内标标准储备液,于-18℃避光保存,有效期18个月。4.12 中间浓度内标溶液:取D4-吡虫啉标准储备液,用甲醇-水溶液(4.5)逐级稀释配制成4µg /mL和200ng/mL,0~4℃保存,有效期6个月。4.13 混合标准工作溶液:准确吸取一定量的中间浓度混合标准溶液(4.10)和中间浓度内标溶液(200 ng/mL),用甲醇-水溶液(4.5)配制成10,20,50,100,200 ng/mL系列浓度的混合标准工作溶液,内标浓度均为20 ng/mL,0~4℃保存,有效期3个月。4.14 微孔滤膜:0.2µm,有机相。4.15 流动相过滤滤膜:0.2µm,水相。5 仪器和设备5.1 高效液相色谱-串联质谱仪(LC-MS/MS):配有电喷雾离子源(ESI)。5.2 电子分析天平:感量分别为0.1 mg和0.01 g。5.3 超声波水浴。5.4 漩涡混合器:3000r/min。5.5 离心机:9000r/min。5.6 离心管:聚四氟乙烯,50mL。5.7 溶剂过滤装置。6 试样的制备和保存6.1 试样的制备与保存取番茄、梨等果蔬样品约500g,将其可食用部分切碎后,用粉碎机粉碎成浆状,混匀,均分成两份作为试样,分装入洁净的容器中,密封。将试样于-18℃以下冷冻保存。取番茄酱样品约500g,混匀,均分成两份作为试样,分装入洁净的容器中,密封。将试样于-18℃以下冷冻保存。取脱水洋葱样品约200g,混匀,均分成两份作为试样,分装入洁净的容器中,密封。将试样于0~4℃保存。注:在制样过程中,应防止样品受到污染或发生农药残留量的变化。7 测定步骤[
本人需要购买甲基汞标准品,不知哪位朋友可以提供有关信息,谢谢!
本人需要购买甲基汞标准品,哪位朋友能提供相关的信息。谢谢!

[img=,671,265]https://ng1.17img.cn/bbsfiles/images/2018/10/201810111004122198_170_3395179_3.png!w671x265.jpg[/img]在GB 2763中甲基托布津的残留物为甲基托布津与多菌灵之和,如果要测甲基托布津的农残,是否多菌灵也要测一下?谢谢
1.PAHs是什么?http://www.liupingnian.cn/wp-content/uploads/2014/06/PAH1.pnghttp://www.liupingnian.cn/wp-content/uploads/2014/06/PAH2.pnghttp://www.liupingnian.cn/wp-content/uploads/2014/06/PAH3.png2.PAHs哪里来?食品中的PAH污染有不同的来源,其中最主要的是环境和食品加工过程的污染。加工过程又被认为是造成食品PAH污染的最主要方式,包括食品的烟熏、烘干、烹饪过程。加工过程的污染的例子:A. 沥青路面晾晒谷物,大豆及各种食品(在中国大部分地区都存在,国外未知)B. 机动车路边等室外晾晒食品(大气中的PAHs吸附在食品上,特别是容易出油的肉,鱼等)C.熏鱼,熏肉等传统加工方法(确实是用烟熏的,现代的加工法中有些只是采用烟熏液,可以显著降低PAHs的污染)D.溶剂提取方式提取食用油(在油桶上的加工方式为浸提),而溶剂会有含有PAHs的风险。E. 厨房油烟,油温太高时,也容易产生多环芳烃。F. 烧烤食品3.PAHs的危害2002年在欧洲,食品科学委员会(SCF)已对33种PAH进行了评价。评价的结果显示在33种PAH中有15种物质在实验动物的体内实验中证明对体细胞具有致突变性和基因毒性,这15种物质分别为苯并蒽,苯并-, -, 荧蒽,苯并二萘嵌苯(benzoperylene),苯并芘,屈(chrysene),环戊芘(cyclopentapyrene), 二苯并蒽(dibenz anthracene),二苯并-,二苯并-,二苯并-,二苯并芘,茚并芘和5-甲基屈。许多具有毒性作用的PAH会在凹陷的区域结合形成复合物。动物实验表明PAH具有多种毒性作用,如血液毒性、生殖和发育毒性及免疫毒性。在低计量时就具有致癌性和致畸性。具有致癌性和致畸性的PAH分子量通常比较大,含有4个或4个以上的环。对于许多PAH,它的潜在的致癌性造成了主要的危害和危险性。已经证实在燃烧过程中产生的许多PAH、煤焦油和包括PAH的各种混合物在实验动物体内、体外的基因毒性和诱变性实验中都显示了致癌性。总的来说,许多证据表明基因毒性与在PAH暴露下DNA加和物形成、突变和癌症发生的致癌机制相似。对实验动物的各种检验结果显示,15种具有基因毒性的PAH,除了苯并 二萘嵌苯外,其余均具有显著的致癌性。虽然只有苯并芘可以使用膳食管理的方法检测,但其他化学物也均被认为对人类具有潜在的基因毒性和致癌性。由于通过饮食摄入PAH对健康有长期副作用,因此将这些化合物列为优先评估的项目进行危险性评估。4. PAHs的限量因为其毒性太大,制定限量的趋势是接近现代最先进仪器的检出限和接近自然本底值。5.PAHs的测试标准中国标准: CJ/T 147-2001城市供水多环芳烃的测定液相色谱法 CNS 15169-2007香品燃烧所产生之多环芳香烃化合物测定法 CNS 15289-2009硫化橡胶制品中加工油多环芳香烃之测定法 CNS 16000-12-2011室内空气-第12部:多氯联苯(PCBs)、多氯二苯并对戴奥辛(PCDDs)、多氯二苯并呋喃(PCDFs)及多环芳香烃(PAHs)之采样策略 GB/T 23213-2008植物油中多环芳烃的测定气相色谱-质谱法 GB/T 24893-2010动植物油脂多环芳烃的测定 GB/T 26411-2010海水中16种多环芳烃的测定气相色谱-质谱法 GB/T 28189-2011纺织品多环芳烃的测定 GB/T 29614-2013硫化橡胶中多环芳烃含量的测定 GB/T 29616-2013热塑性弹性体多环芳烃的测定气相色谱-质谱法 GB/T 29670-2013化妆品中萘、苯并蒽等9种多环芳烃的测定气相色谱-质谱法 GB/T 29784.1-2013电子电气产品中多环芳烃的测定第1部分:高效液相色谱法 GB/T 29784.2-2013电子电气产品中多环芳烃的测定第2部分:气相色谱-质谱法 GB/T 29784.3-2013电子电气产品中多环芳烃的测定第3部分:液相色谱-质谱法 GB/T 29784.4-2013电子电气产品中多环芳烃的测定第4部分:气相色谱法 GBZ/T 160.44-2004工作场所空气有毒物质测定多环芳香烃类化合物 HJ 478-2009水质多环芳烃的测定液液萃取和固相萃取高效液相色谱法 HJ 646-2013环境空气和废气气相和颗粒物中多环芳烃的测定气相色谱-质谱法 HJ 647-2013环境空气和废气气相和颗粒物中多环芳烃的测定高效液相色谱法[table=100%,tra
最近要测n甲基甲酰胺标准品但是找了一些标准品厂家都没这个标,要证书的,所以也不能用溶剂代替。有买过的或者有货号的请提供一下给我
最近在做塑料制品中的18种多环芳烃,5ug/mL浓度(为了确定保留时间所以浓度较高)的峰形都很好。配制了20ng/mL~400ng/mL共6个浓度点的标准溶液,出来的谱图无一例外表现出:除了前面3个化合物外,其余15种都拖尾严重,挨着的2个或3个化合物以一个峰宽很大的峰出来,而且保留时间都比5ug/mL时延迟。奇怪的是,我做了中间浓度点的加标实验,样液中的一系列化合物峰形都很正常。标准物质用甲苯配制,塑料制品用甲苯提取。 也就是说,标准物质在溶剂中拖尾严重,在样液中峰形正常。这是怎么回事? 我老化了柱子还是如此。望高人指点。
谁知道羧甲基淀粉钠应用于食品的标准?
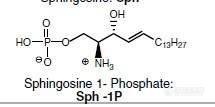
http://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471106_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471108_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471109_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152016_471110_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152016_471111_2803904_3.jpg为什么标准品浓度不一样?峰型也不一样? 峰拖尾严重如何处理?标准品怎么会有两个峰?我的用的是C18拄,色谱条件是甲醇:水(0.1%甲酸,10mm的乙酸铵90:10,等度洗脱。做所得是2种磷脂分子,分子式如上
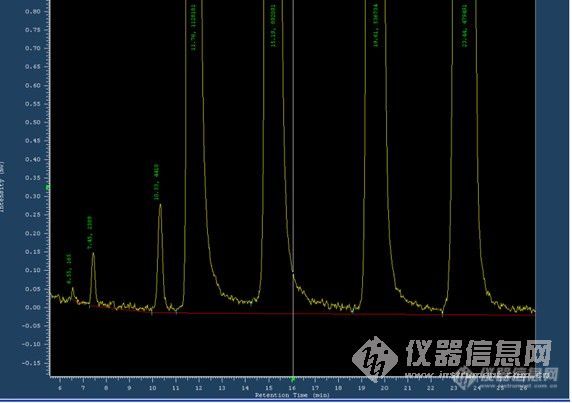
各位好心人,最近我在做饲料当中一类药物的质量检测标准,以下是用标准品进的样,不放大看峰形比较好,放大后就不好了,请帮忙看看拖尾严重吗?符合质量标准的要求吗?另外,在做这一类质量标准时对于拖尾有没有什么要求呢?四个峰分离效果怎么样呢?谢谢好心人了!![img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003032229_203578_1638724_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003032229_203579_1638724_3.jpg[/img]
[align=left][/align]【作者】:崔靖 韩冬梅 徐隆昌 韦薇【题名】:曲妥珠单抗生物类似药质量"相似性评价标准"探讨【期刊】:药学学报【年、卷、期、起止页码】:2021-01-01【全文链接】:10.16438/j.0513-4870.2021-0998
最近检测样品收样要求检甲基托布津,查资料发现甲基托布津遇水就变成多菌灵了,那测甲基托布津有意义吗?请高手指点
需要购买氯化甲基汞、氯化乙基汞的纯品标准,请提供相关的信息及联系方式。
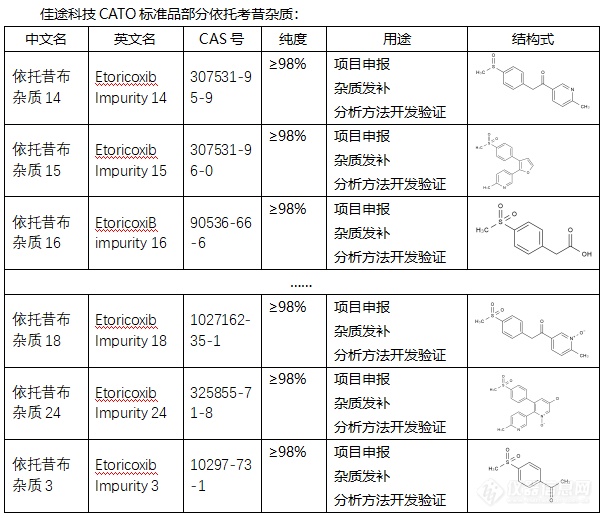
[font=宋体]◇依托考昔杂质[/font][font=宋体][font=宋体] 依托考昔杂质是在依托考昔的生产或保存过程中产生的非目标化合物。这些杂质可能会影响依托考昔的纯度和药效,因此在依托考昔的生产和质量控制过程中需要严格控制其含量。依托考昔杂质有多种类型,每一种都具有不同的化学特性,如[/font][font=Calibri]CAS[/font][font=宋体]号、分子式、分子量等。例如,依托考昔杂质[/font][font=Calibri]K[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]349536-41-0[/font][font=宋体],分子式为[/font][font=Calibri]C18H15ClN2O3S[/font][font=宋体]。依托考昔杂质[/font][font=Calibri]Etoricoxib[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]202409-33-4[/font][font=宋体],分子式为[/font][font=Calibri]C18H15N2O2SCl[/font][font=宋体],别名包括依托考昔、[/font][font=Calibri]5-[/font][font=宋体]氯[/font][font=Calibri]-2-(6-[/font][font=宋体]甲基吡啶[/font][font=Calibri]-3-[/font][font=宋体]基[/font][font=Calibri])-3-(4-[/font][font=宋体]甲基磺酰苯基[/font][font=Calibri])[/font][font=宋体]吡啶等。依托考昔杂质[/font][font=Calibri]Q[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]292067-97-1[/font][font=宋体],分子式为[/font][font=Calibri]C18H15ClN2S[/font][font=宋体]。此外,依托考昔还可能存在其他未具体命名的杂质。[/font][/font][font=宋体][font=Calibri] CATO[/font][font=宋体]标准品提供的依托考昔全套的杂质[/font][/font][font=宋体],[/font][font=宋体]这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分[/font][font=宋体]。[img=,604,518]https://ng1.17img.cn/bbsfiles/images/2024/02/202402182043373327_8351_6381607_3.png!w604x518.jpg[/img][/font][font=宋体][color=#05073b][back=#fdfdfe] 广州[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]佳途科技[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]股份有限公司[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]深知药物研发与质量控制的重要性[/back][/color][/font][font=宋体][font=宋体],[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供依托考昔全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展[/font][font=宋体],[/font][font=宋体]以满足客户在药物研发和质量控制方面的需求。[/font]
甲苯中甲基汞的标准品用GB5009.14法检测,标准品用什么溶剂溶解?望老师不吝赐教
食品安全国家标准 食品营养强化剂 L-硒-甲基硒代半胱氨酸
求购棒曲霉素和5-羟甲基糠醛标准品!!!我的联系方式:niupf0406@163.com

【作者中文名】张亮; 道毅俊; 李中东; 张怡; 施孝金; 钟明康;【作者英文名】ZHANG Liang DAO Yi-jun LI Zhong-dong~* ZHANG Yi SHI Xiao-jin ZHONG Ming-kang(Department of Pharmacy; Huashan Hospital; Fudan University; Shanghai 200040; China);【作者单位】复旦大学附属华山医院药剂科;【摘要】目的建立HPLC荧光检测法测定曲马多及活性代谢物O-去甲基曲马多在血浆中的浓度,研究曲马多片在健康人体中的药动学及生物等效性。方法采用随机交叉自身对照试验设计,20名健康男性受试者单次po 100 mg曲马多口腔崩解片和曲马多片后,按规定时间采集肘静脉血,血样经液.液萃取处理,以0.03 mol·L~(-1)四硼酸钠(含0.5%三乙胺,磷酸调pH至4.0)-甲醇(75:25)为流动相,色谱柱为Diamonsil C_(18)柱(4.6 mm×200mm,5μm),流速1.0 mL·min~(-1),柱温50℃,荧光检测的激发波长275 nm,发射波长304 nm,测定曲马多和O-去甲基曲马多的血药浓度,并计算两制剂的主要药动学参数及相对生物利用度。结果测定血浆中曲马多和O-去甲基曲马多的最低检测限均为1.0μg·L~(-1),曲马多和O-去甲基曲马多分别在1.0~600.0和1.0~300.0μg·L~(-1)内线性关系良好;血药浓度测定的日内、日间精密度RSD均小于5%;测得曲马多口腔崩解片和曲马多片的血样中曲马多的主要药动学参数为:t_(max)(2.2±1.0)和(1.9±0.9)h,ρ_(max) (...http://ng1.17img.cn/bbsfiles/images/2012/08/201208271734_386591_2379123_3.jpg

大神们,求助:为什么直接进苯的标准品峰形很好,但是热解析进样是拖尾的?第一张热解析进样,第二张直接进样甲醇中苯,http://ng1.17img.cn/bbsfiles/images/2016/12/201612281625_02_2196181_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281626_01_2196181_3.jpg
艾曲波帕杂质是一种化学物质,它是艾曲波帕的同分异构体或相关化合物。艾曲波帕是一种血小板生成素受体激动剂,用于治疗慢性免疫性血小板减少症。COTO标准品是一种高纯度的标准物质,用于测定艾曲波帕及其杂质的纯度、含量和化学性质。通过与COTO标准品进行对比和分析,可以确定艾曲波帕及其杂质的结构、组成和含量,从而保证艾曲波帕的质量和安全性。在药物研发和生产过程中,COTO标准品的使用非常重要。它可以提供可靠的参照物,用于质量控制、药物分析和化学计量学研究。通过使用COTO标准品,可以确保艾曲波帕及其杂质的准确性和可靠性,为药物的安全性和有效性提供保障。总的来说,COTO标准品在艾曲波帕杂质的研究和控制中具有重要作用。通过使用COTO标准品,可以更好地了解艾曲波帕及其杂质的性质和含量,从而确保药物的安全和有效性。同时,也需要加强生产过程中的管理和监督,加强质量标准和监管措施的执行力度,确保药物质量和安全。
采用液液萃取的方法测三卤甲烷,做出了标准品的曲线,然后用去离子水做了一个加标的,测了发现加标的峰面积比标准品的峰面积大很多,是不是很不正常?这怎么计算回收率?而且萃取不是会有损失,怎么峰面积还变大了?前处理步骤,就是20ml水样加入比色管中,然后加100ppb的标准品,加4ml的甲基叔丁基醚萃取,再加入8g的无水硫酸钠。也做了个空白,发现空白没有这些出峰,峰面积大小可以忽略不计。加标和测标准品的方法是一样的。请高手解答一下
文献上用亚甲基蓝脱色力来衡量活性炭的能力,想问一下甲基蓝脱色力是不是就是标准上说的甲基蓝吸附量?如果不是,那它是什么?具体怎么算一直弄不明白谢谢了
工程院院士陈君石:中国食品标准就是妥协产物作者:吕宗恕字号:陈君石——访国家食品安全标准审评委员会副主任委员陈君石某种程度上说,没有妥协,就没有办法制定标准。很多道理在全世界行得通,但在中国就行不通。76岁的陈君石又被“骂”了。一周前还躺在病床上的这位中国工程院院士,在蒙牛黄曲霉素超标处于群情激愤时刻,还是忍不住要发言。他通过网络提醒公众,“对于此次事件,公众没有必要惊慌,因为大家不会天天吃到含黄曲霉素的食品。” 此言刚出,网上一片骂声。这已经不是他第一次挨骂了。“阜阳奶粉没有有毒有害物质,它只造成了人体健康的影响”、“我们政府对食品安全的监管力度在全世界绝对是第一”……近年多起食品安全事件中,这个直言不讳的老头屡屡被骂,就连他身边的朋友也有耳闻。不过,在一些人眼中,他的这些话不过是对常识的重申。陈君石]中国食品[/url][5.85 -0.34%]毒理学学科的创始人之一,中国疾控中心营养与食品安全所研究员。他还是国家食品安全风险评估专家委员会主任委员、国家食品安全标准审评委员会副主任委员,此外,他还在国际食品添加剂法典委员会等多家权威机构兼任要职。面对公众和媒体对食品标准一波又一波的“讨伐”,半躺在病床上的陈君石细心回答南方周末记者的每一个问题,他说,“别人有顾忌,但我没有,该讲的还得讲,哪怕有骂名。” 国标不是落后,而是矛盾易见南方周末:您参加了2011年12月初召开的食品安全国家标准审评委员会第六次主任会议,这次会议研究了食品安全国家标准规划(2011-2015)征求意见稿,可否谈下该规划的出台背景?陈君石:背景很重要。在食品安全法颁布之前,中国是世界上唯一有多套国家级食品标准的国家,这种现象只有中国有。就同一食品而言,根据食品卫生法,有食品卫生标准,主管部门是卫生部;根据产品质量法,有产品质量标准,主管部门是国家质检总局;根据研报[/url]]质量安全法,有农产品质量安全标准,主管部门是农业部。这三套标准都具有国家强制性,其间矛盾显而易见,企业苦不堪言。因为不是一个部门制定的,互相不通气,在安全指标这一共同指标上,比如,同一食品测定铅含量,若按这个标准是合格,按那个标准就不合格了。所以,2009年颁布的食品安全法规定,今后我们国家只有一套国家级强制性的食品安全标准。也就是说,质量的指标不纳入国家食品安全标准体系。从2009年开始,国家开始清理整顿。南方周末:两年多来,这项清理整顿工作进展如何?遵循原则是什么?遇到的最大困扰又是什么?陈君石:按照工作步骤,以标准审评专业委员会、大委员会(指标准制定中的分级统筹),到卫生部层面,一步一步地推进中。遵循原则是只有一套国家强制性食品安全标准,整顿过程中最大的困难就是清理整顿标准中,各部门、各位专家之间的看法不一,存在分歧,不过,最后总能通过妥协达成一致意见。南方周末:公众喜欢拿中国食品标准和国外相比,有时会得出“落后”等评价,你如何评价?陈君石:我不认为中国标准是落后的,国与国之间的情况不同,食品标准不尽相同,不能简单、机械化地比较。标准不是越高越好,关键在执行。简单来说,我国的食品标准总体上是适用的。修订前存在多套标准,修订时参照国外标准,兼顾本国国情,这是世界通行做法。南方周末:比如乳品新国标,媒体和公众都说是二十年来的倒退,为什么?陈君石:倒退不是事实。与过去相比,乳品新国标突出与人体健康相关的限量规定以及标准的强制性,其中对餐桌上可见产品的各项指标均有提高。以婴儿配方食品为例,新国标增加了十多项检测内容,有些标准比国际标准还严格。在新国家食品安全标准中一般不包括质量指标,但并不等于标准中不能有质量指标或标准。如根据质量,将产品分成一、二、三级,但与食品安全没有关系,不能说一级就是安全的,二级就不安全,三级就更差劲了。没有妥协,就没有办法制定标准南方周末:乳品新国标出台之前,各方争执不休,这些争执有无依据?陈君石:两年多来,新乳品标准修订至少开了五六十次各种各样的专家会议,还不包括领导阶层的会,争得不亦乐乎。我不理解为什么生鲜乳要规定蛋白质含量。蛋白质多一点少一点跟食品安全有什么关系?一个是每百克鲜乳蛋白质含量2.8克,一个是2.95克,也就是每百克鲜乳蛋白质含量差0.15克,喝500克牛奶也差不了一克蛋白质。而我们通常一天三餐要吃70克蛋白质,我们讨论这不到一克蛋白质的问题有什么意义?乳品新国标一共公布了66个标准,真正引起广泛争论的只有其中一个标准的两个指标,即生鲜乳标准中的蛋白质含量和菌落总数。就算这两项落后的话,也不能判定我们这个标准就是全世界最落后的标准。还有几百项指标不落后,为什么就没人说呢?南方周末:您的意思是反对者是以点代面,不看整体水平?陈君石:是的。在制定乳品标准过程还有一个争论是要不要制定生鲜乳的标准,我们搞公共卫生的人认为没有必要。因为谁喝生鲜奶?收购时生鲜乳蛋白质含量是每百克鲜乳蛋白质含量2.8克,但最终产品的蛋白质含量多少取决于厂家,厂家根据不同的产品来规定不同的蛋白质含量。南方周末:您怎么看标准制定中的妥协问题?陈君石:某种程度上说,没有妥协,就没有办法制定标准。标准是一个妥协的产物。很多道理在全世界行得通,但在中国就行不通。有的部门、专家同意,可是别的部门、专家就不同意,一定要制定生鲜乳的标准。这个乳品标准来回讨论不知道多少次了,最后只有妥协。南方周末:公众对企业参与标准制定意见也很大,如何来理性对待?陈君石:公众是被媒体误导了。起草某些标准需要企业参与,因为企业不仅最了解生产情况,也是标准的执行者,如果一套标准出台后,超过50%的样品不合格,那这套标准还有何意义?在整个国家食品安全标准的制定过程中,企业只参加起草,并不参与评审和最后的卫生部行政审查。食品安全不可能零风险南方周末:乳品新国标争议未了,即将实施的速冻食品新国标再起波澜,这种争议背后有什么社会背景?陈君石:速冻食品中的金黄色葡萄球菌不是最要命的最厉害的致病菌,沙门氏菌、李斯特氏菌等比它厉害多了,尽管葡萄球菌靠后,但是原有标准规定葡萄球菌不得检出是非常不科学的。什么叫食品安全?食品安全就是讲风险,不得检出在国际上是一个不科学的做法,这次,我更不明白,为什么公众说这个标准倒退了?现在我们的做法是,五个样品允许有一个样品呈阳性,而且还有一定的细菌量,这不是中国人的发明,只不过跟国际接轨而已。南方周末:这是不是说,公众得到的信息容易出现偏差?陈君石:对,这就是我过去讲到的我们食品安全是没有零风险的。你今天来采访我就没风险吗?当然有风险,坐飞机、打出租也有风险。但部分公众没有明白其中的道理,只看到数字的高低,以此来判断倒退与否。南方周末:您认为应该如何避免上述争议的出现?陈君石:一边倒声音出现后,政府应该设有专门部门、人员及专门经费来沟通,这称之为风险交流。这不是信息发布,信息发布仅仅是风险交流的一部分。现在,政府往往反应滞后,透明度不够,话也讲得不够多,因为讲多了怕出毛病。此外,社会还缺少一个风险交流的平台——一个民间的,能专门提供科学知识的平台。其实,中国每年因食品安全事故死的人数并不多,国务院为什么唯独要成立食品安全委员会,而不成立车祸委员会、癌症委员会或心脏病委员会呢?因为食品安全远远不是公共卫生问题,而是社会问题、政治问题,甚至是社会安定团结问题。
求助:以下标准ASTM D 1983 甲基酯气液色谱法分析脂肪酸组分的试验方法ASTM D 1984 妥尔油脂肪酸ASTM D 3457 气态-液态色谱法测定脂肪酸组分,从脂肪酸制备甲基酯的试验方法ASTM D 1537 蒸馏的大豆脂肪酸ASTM D 1538 蒸馏的亚麻籽脂肪酸ASTM D 1539脱水蓖麻籽脂酸
腐乳中二甲基黄和二乙基黄的测定解决方案二甲基黄和二乙基黄,属于工业染剂,主要用于石蜡、塑胶、印刷油墨、石油和肥皂等的着色,具有致癌性。因其不属于食品添加剂范畴,从未列入台湾监控部门的常规检查项目,被当地不法商人利用至今。目前,关于该类物质的测定方法几乎没有报道。方法优势:迪马科技建立固相萃取-超高效液相色谱串联质谱法同时检测腐乳中二甲基黄和二乙基黄,本方案具有以乙腈为提取液,采用ProElut DYC固相萃取柱净化样品,通过UPLC- MS/MS检测;前处理步骤简单、净化效果好、回收率高、基质效应小优点;保证实验结果准确性、重现性。方法检出限0.03 μg/kg,定量限为0.1 μg/kg;适用于各省市出入境、质检、疾控、食品药品检验所、第三方检测机构、食品检测机构等。专用柱优势:ProElut DMY 柱由2种吸附剂按照一定的比例分层填装而成,采用不同作用机理去除杂质,同时对二甲基黄和二乙基黄没有不可逆吸附,保证了样品的净化效果及回收率;本产品是商品化的成品柱,不用手工填装,吸附剂稳定性好,不受外界环境因素影响,保证实验结果的重现性和准确性;过柱过程操作步骤简单,节省时间,提高了工作效率以下为详细解决方案,敬请参考!腐乳中二甲基黄和二乙基黄的测定1、适用范围 适用于腐乳中二甲基黄和二乙基黄的检测,二甲基黄的方法检出限是0.03 μg/kg,二乙基黄的方法检出限是0.04 μg/kg,定量限是0.1 μg/kg。2、提取取1.0 g样品,加1.0 g氯化钠与5 mL乙腈,涡旋混匀,6000 rpm下离心2 min,精密量取2.5 mL上清液待净化。3、净化——ProElut DMY 3 mL(Cat#:65914)a活 化:3 mL乙腈活化; b上 样:c淋 洗:加入待净化液,弃去流出液;加入3 mL乙腈,弃去流出液;d洗 脱:加入4 mL10%氨水甲醇,收集流出液;d重新溶解:将流出液在50 ℃下氮吹至干,用乙腈定容至1 mL,过0.22 μm微孔滤膜,供LC-MS分析。4、分析条件4.1 UPLC 条件:色谱柱:Endeavorsil C18,100 × 2.1 mm,1.8 μm (Cat.# 87003)流 速:0.2 mL/min进样量:5 μL柱 温:35 ℃流动相: A:0.1%甲酸水 B:乙腈梯度设置时间/Min.055.510A(%)20102020B(%)809080804.2 质谱条件:电离模式:ESI扫描方式:正离子扫描检测方式:多反应监测电喷雾电压:5500 V雾化气压力:50 psi辅助气压力:50 psi气帘气压力:20 psi离子源温度:500 ℃定性离子对、定量离子对、碰撞气能量及去簇电压见下表药物名称定性离子对定量离子对碰撞气能量/eV去簇电压/ V(m/z)(m/z)(母离子/子离子)(母离子/子离子)二甲基黄226.2/77.0226.2/77.02877226.2/134.128二乙基黄254.2/120.2254.2/120.23473254.2/148.126254.2/134.1335、添加回收结果腐乳中二甲基黄和二乙基黄的LC-MS检测添加回收结果分析物基质添加水平(μg/kg)回收率(%)二甲基黄黄色腐乳1.095.4二乙基黄1.098.6二甲基黄红色腐乳1.090.5二乙基黄1.092.3http://www.dikma.com.cn/u/image/2016/01/29/1454056478555325.jpg二甲基黄标准多反应监测色谱图http://www.dikma.com.cn/u/image/2016/01/29/1454056486222259.jpg二乙基黄标准多反应监测色谱图http://www.dikma.com.cn/u/image/2016/01/29/1454056492122223.jpg添加水平为1.0 μg/kg黄色腐乳中二甲基黄和二乙基黄检测的多反应监测色谱图[/alig
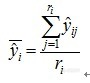
[align=center][b]基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的2,3,5-三甲基苯醌粗品萃取过程定量模型优化研究[/b][/align][b]中文摘要:目的[/b]实际工业生产工艺中,萃取是一项耗时耗力的过程,萃取终点的确定通常采用离线的HPLC, [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]或由熟练工人根据经验判断,这些方法操作较复杂或是不够准确,在实际生产中缺乏一种快速有效的检测手段以判断萃取终点,节省操作时间,避免过分萃取浪费溶剂。利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术可以明显改善萃取工艺。[b]方法[/b]本实验针对2,3,5-三甲基苯醌(TMBQ)粗品萃取环节,采用偏最小二乘法(PLS)建立模型,考察了不同预处理方法与变量选择方法对模型的影响以优化模型,采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]结合PLS算法建立TMBQ萃取过程含量快速检测模型,并使用不同预处理方法与波段选择方法对模型进行优化,最终确定使用一阶导数+SG15点平滑预处理结合iPLS选择波段建立PLS模型。[b]结果[/b]建立模型的各项参数为:波普区间4385.33cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup], 5928.11cm[sup]-1[/sup]-6309.94cm[sup]-1[/sup],模型决定系数R[sup]2[/sup]=0.996, RMSEP=0.1350。[b]结论[/b]建立的模型精密度与准确度良好,可以满足含量分析的需要,是TMBQ萃取过程含量快速检测的有效方法,可以快速准确的对三甲基苯醌粗品萃取过程进行在线监测,提供了一种用于该工艺环节的快速检测手段,如果应用于生产,可以节省操作时间,避免溶剂浪费。[b]关键词:[/b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析;2,3,5-三甲基苯醌;萃取 2,3,5-三甲基苯醌是维生素E的主要中间体。2,3,5-三甲基苯醌在国外已有生产, 但国内尚未见文献报道。国内用2,3,5-三甲基苯醌主要依赖进口。因此,开展2,3,5-三甲基苯醌的合成研究对发展国内维生素 E 的生产具有重要意义。TMHQ的合成工艺国内外己有多种报道,较为先进的是TMP法与异佛尔酮法,TN[b]B[/b]Q粗品萃取过程是合成TMBQ的关键环节。在制药领域,NIRS作为一种重要的PAT工具,已成功用于药物的原辅料评价、关键过程的监测和控制、成品的快速放行和质量监测等各个环节,为保证产品质量、降低生产成本、革新生产过程发挥了重要的作用。[b]1实验材料与仪器1.1仪器[/b] Antaris Ⅱ傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国Thermo Fisher公司),7890A[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]-氢焰离子化检测器(美国Agilent公司),HP-1毛细管色谱柱(美国Agilent公司)BT224S电子分析天平(德国Sartorius公司),容量瓶,100ml圆底烧瓶,分液漏斗,[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url][/color][/url](美国ThermoFisher公司)。RESULT[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]采集软件,TQAnalyst[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析软件,Matlab数据处理软件。[b]1.2试剂[/b] 2,3,6-三甲基苯醌(合成步骤见第二章),石油醚(天津富宇精细化工有限公司,沸程60℃-90℃)。[b]2方法2.1样品制备和处理[/b] 按照第二章步骤合成得TMBQ得其石油醚溶液,萃取水相合并有机相,旋蒸浓缩除去石油醚至橙黄色油状液体,称重,再用石油醚作为溶剂配置1ug/ml~50mg/ml一系列溶液。[b]2.2光谱采集[/b] 波长范围4000 cm[sup]-1[/sup]-10000cm[sup]-1[/sup];扫描次数32;分辨率8 cm[sup]-1[/sup],使用4mm光程的玻璃样品管乘装液体样品,采集样品前采集背景以消除背景干扰,每个样品重复采集三次光谱。光谱采集在恒定室温(24℃)与恒定湿度的条件下进行。[b]2.3样品集划分[/b] 使用K-S分类法将所有66个样品换分为48个校正集与18个验证集。[b]2.4模型建立与优化[/b] 采用导数、平滑等方法对原始光谱进行预处理,应用偏最小二乘法(PLS)建立模型,结合RMSEP等评价参数,通过变量选择方法选择特征波段优化模型。[b]2.5 重复性考察[/b] 选择3个验证集样品,每个样品连续采集10次光谱,使用建立好的模型预测每张光谱,并计算出每个样品十次预测值的均值和标准偏差。是第i个样品的第j张光谱,第i个样品共测定ri个光谱,第i个样品的预测平均值为:[align=center][img=,90,83]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311044_01_1626619_3.png[/img][/align] 复测定的标准偏差为:[align=center][img=,164,102]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311044_02_1626619_3.png[/img][/align] 用c[sup]2[/sup]检验来考察这些重复性标准偏差是否属于同一总体:[align=center][img=,271,245]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311045_01_1626619_3.png[/img][/align] z为需要重复测定的样品数,将所得χ[sup]2[/sup]与自由度(z-1)临界值比较,若χ[sup]2[/sup]在临界值以下,则重复测定的所有方差属于同一总体,标准偏差均值σ可以作为近红外测定的标准偏差,近红外分析方法的重复性为z××σ[sub]max[/sub]。如果χ[sup]2[/sup]大于临界值,近红外分析方法的重复性随样品组分浓度不同而不同,这时,近红外分析方法的重复性不大于z××σ[sub]max[/sub](σ[sub]max[/sub]为σi中的最大值)。[b]2.6[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测[/b] 初始温度180℃恒温5min,以10℃/min的速率升温至240℃。进样口温度:300,检测器温度:300,载气:氮气,载气流速:3ml/min,进样量:0.5ul。[b]3结果3.1校正集与验证计划分[/b] 使用K-S分类法将所有66个样品换分为48个校正集与18个验证集。校正集与验证集的第一第二主成分分布图如图1,其中黑色符号代表校正集样品,红色符号代表验证集样品,验证集均匀分布于校正集中,可见使用该方法分类合理。[align=center][img=,553,217]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311047_01_1626619_3.png[/img][/align][align=center]图1 所有样品主成分分布图[/align][b]3.2预处理方法的选择[/b] 考察无预处理、一阶导数+SG5点平滑、一阶导数加SG9点平滑、一阶导数+SG15点平滑、二阶导数加15点平滑这几种方式的建模结果,以RMSEC、RMSECV、RMSEP以及R[sup]2[/sup]作为评价指标,结果见表1。[align=center]表1 预处理方法评价参数[/align][align=center][img=,566,164]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311104_01_1626619_3.png[/img][/align] 无预处理的模型结果最差,说明噪声对模型结果有较大影响,原始光谱如图2。SG15点平滑+一阶导数的预处理结果RMSEC、RMSECV以及RMSEP最小,R[sup]2[/sup]最高。因此选择SG15点平滑+一阶导数作为模型的预处理方法,预处理后光谱如图3。[align=center][img=,524,224]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311048_01_1626619_3.png[/img][/align][align=center]图2 原始光谱图[/align][align=center][img=,532,210]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311049_01_1626619_3.png[/img][/align][align=center]图3 一阶导数+SG15点平滑预处理光谱图[/align][b]3.2异常样本的剔除[/b] 图4为校正集样品在学生残差-杠杆值图中的分布。图中5号(红色方框标记)样品学生残差值与杠杆值都非常高,判定为异常样品,猜测为溶液配制错误或者在光谱采集过程中出现错误,因此在后期模型优化中剔除这一异常值。[align=center][img=,563,217]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311050_01_1626619_3.png[/img][/align][align=center]图4 学生残差-杠杆值关系图[/align][b]3.3波段选择结果[/b] 以一阶导数+SG15点平滑为最优预处理方法进行波段选择,主要考察ForwardiPLS、SPA、相关系数法三种方法。[b]3.3.1iPLS波段选择结果[/b] 设定20为最大主成分数,分别考察以50、100、200个变量为波段基础的建模效果。红色虚线是全波段建模的RMSECV,红色与绿色条带的高度代表以此条带的变量建模所得RMSECV,从图5中可见,绿色条带的RMSECV值最小,因此绿色条带是被选择用于建模的波段,红色条带则表示不被选择的区域。表2为各变量基础的模型参数。[align=center][img=,558,268]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311051_01_1626619_3.png[/img][/align][align=center]图5 以50个变量为基础的iPLS法波段选择效果图[/align][align=center][img=,572,266]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311052_01_1626619_3.png[/img][/align][align=center]图6 以100个变量为基础的iPLS法波段选择效果图[/align][align=center][img=,618,262]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311052_02_1626619_3.png[/img][/align][align=center]图7 以200个变量为基础的iPLS法波段选择效果图[/align][align=center]表2 不同变量基础的建模结果[/align][align=center][img=,646,111]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311053_01_1626619_3.png[/img][/align][b]3.3.2 SPA法波段选择结果[/b] SPA算法首先通过完成n个波长分组各M个波长选择,然后通过多元定量校正模型完成m(1£m£M)个最优波长的选定。图8为SPA法选择变量的效果图。 运行SPA算法共选择3个变量,对应波数为4188.65cm[sup]-1[/sup],4885.50cm[sup]-1[/sup],7503.50cm[sup]-1[/sup],为图中红色方框标注,以此3个变量建立PLS模型,结果如表 所示,RMSECV与RMSEP均有所增加,R[sup]2[/sup]降低,表明模型预测能力与线性都有所降低。分析原因可能是此方法在选择波段过程中由1557个变量减少到3个,光谱变量删除过多,去除大量无关变量的同时导致许多有价值信息的丢失。[align=center][img=,501,246]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311053_02_1626619_3.png[/img][/align][align=center]图8 SPA算法变量选择结果图[/align][b]3.3.3相关系数法波段选择结果[/b] 将相关系数阈值设定为0.6、0.7、0.8,使用相关系数法计算出TMBQ含量值与波数的相关系数图,如图9,图中虚线为设定的相关系数阈值,虚线以上及以及的部分代表相关系数大于阈值的波段,阈值越高,被选择的波段越少,当阈值设为0.8时,大于阈值的波段已经较少。以超过阈值的波段建立PLS模型。模型结果如表3,可见将阈值设为0.6时模型结果最好。[align=center] a[img=,402,175]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311055_01_1626619_3.png[/img][/align][align=center] b[img=,409,187]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311056_01_1626619_3.png[/img][/align][align=center] c[img=,409,176]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311056_02_1626619_3.png[/img][/align][align=center]图9 不同阈值的波数相关图(a阈值设为0.6,b阈值设为0.7,c阈值设为0.8)[/align][align=center]表3 相关系数法建模参数[/align][align=center][img=,496,105]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311058_01_1626619_3.png[/img][/align][b]3.4 小结[/b] 综合比较全波段建模与三种波段选择方法建模结果,参数如表。其中使用iPLS法选取600个变量,波段区间为4385.33cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup],5928.11cm[sup]-1[/sup]-6309.94 cm[sup]-1[/sup],分别对应双键上C-H第一组合频与一级倍频吸收,建模后具有最高的决定系数和最低的各项方差值,这些参数表明使用该方法建立的模型预测能力最好,与真实值最接近。因此本实验主要选择iPLS方法选择变量,结合一阶导数+SG15点平滑建立模型,应用于TMBQ萃取过程含量的快速检测。[align=center]表4 各变量选择方法比较[/align][align=center] [img=,374,136]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311059_01_1626619_3.png[/img][/align][align=center][img=,524,214]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311059_02_1626619_3.png[/img][/align][align=center]图10 优化后模型预测线性图[/align][b]3.5重复性试验考察[/b] 采集验证集8号、25号、36号样品,对TMBQ含量模型进行重复性测试,每样品采集10次光谱。预测结果见表5。[align=center]表5 重复性考察结果[/align][align=center][img=,578,337]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311100_01_1626619_3.png[/img][/align] 自由度为2时,χ[sup]2[/sup]临界值为5.99。实际χ[sup]2[/sup]小于临界值,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析方法重复性为0.154,可以满足分析应用。[b]3.6NIR预测考察[/b] 第一次使用20ml石油醚萃取,之后每次使用等体积10ml石油醚萃取,共萃取8次,使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]测定TMBQ峰面积,并使用NIR采集8次萃取液[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url],使用优化好的定量模型对其含量进行预测。[align=center][img=,490,255]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311102_01_1626619_3.png[/img][/align][align=center]图11 NIR预测值[/align] 图11为NIR对萃取过程的预测结果,第一次萃取即将大部分产品萃取出,随后的每次萃取量呈逐渐下降的趋势,在第五次萃取后,萃取液中产品含量几乎为0,并且随后没有变化,表明已达到萃取终点。使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测第4~8次萃取液,记录TMBQ峰面积,结果如表6。[align=center]表6 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测结果表[/align][align=center][img=,529,66]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311103_01_1626619_3.png[/img][/align] 第五次萃取后,TMBQ峰面积已经很小,并且基本没有变化,因此在4次萃取完全可以将水相中的TMBQ萃取完全,继续萃取已经没有意义,[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测与NIR预测结果相符,表明此模型预测能力良好,对萃取工艺具有一定指导意义。[b]4讨论[/b] 本实验采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]结合PLS算法建立TMBQ萃取过程含量快速检测模型,并使用不同预处理方法与波段选择方法对模型进行优化,最终确定使用一阶导数+SG15点平滑预处理结合iPLS选择波段建立PLS模型,建模所用波段区间为4385.33 cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup],5928.11 cm[sup]-1[/sup]-6309.94cm[sup]-1[/sup],模型决定系数R[sup]2[/sup]=0.996,RMSEP=0.1350。使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]验证了NIR模型对萃取过程与终点的预测能力。以上结果表明模型精密度与准确度良好,可以满足含量分析的需要,是TMBQ萃取过程含量快速检测的有效方法。[b]5参考文献[/b]孙月婷. 维生素E 的合成与分析研究现状. 广州化工, 2011, 39(6): 34-35.O.A.Kholdeava Synthesis of Vitamia E J.Mol.Cotal,1992,88(5):235~ 244孔黎明, 周涛, 菅盘铭. 2, 3, 5- 三甲基苯醌和2, 3, 5- 三甲基氢醌的一种合成方法: 中国, 102219665. 2011-10-19.A BShishmakov, Yu V Mikushina, O V Koryakova. Oxidation of 2,3,6-Trimethylphenolon Titanium Dioxide Xerogel by Hydrogen Peroxide in the Absence of an OrganicSolvent. RUSSIAN JOURNAL OF APPLIED CHEMISTRY, 2011, 84(9):1555-1559. O V Zalomaeva, N N Trukhan,I D Ivanchikova, et al. EPR study on the mechanism of H[b][sub]2[/sub][/b]O[b][sub]2[/sub][/b]-basedoxidation of alkylphenols over titanium single-site catalysts. J. Mol.Catal. A: Chem., 2007, 277(1-2), 185~192.褚小立. 化学计量学方法与分子光谱分析技术.北京 化学工业出版社. 2011.董学锋,戴连奎,黄承伟等.结合PLS-DA与SVM的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]软测量方法
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7661700512 色氨酸杂质A Tryptophan Related Compound A 对照品/标准品1700501 L- 色氨酸 L-Tryptophan 对照品/标准品1700002 结晶胰蛋白酶 Trypsin Crystallized 对照品/标准品1699005 托吡卡胺 Tropicamide 对照品/标准品1698002 氨丁三醇 Tromethamine 对照品/标准品1697000 醋竹桃霉素 Troleandomycin 对照品/标准品1696958 三乙醇胺 Trolamine 对照品/标准品1696200 三水杨酸 Trisalicylic Acid 对照品/标准品1696109 盐酸曲普利啶 Z- 异构体 Triprolidine Hydrochloride Z-Isomer 对照品/标准品1696007 盐酸曲普利啶 Triprolidine Hydrochloride 对照品/标准品1695004 盐酸曲吡那敏 Tripelennamine Hydrochloride 对照品/标准品1693009 三甲沙林 Trioxsalen 对照品/标准品1692710 三甲丙咪嗪杂质A Trimipramine Related Compound A 对照品/标准品1692709 三甲丙咪嗪马来酸盐 Trimipramine Maleate 对照品/标准品1692527 甲氧苄啶杂质B Trimethoprim Related Compound B 对照品/标准品1692516 甲氧苄啶杂质A Trimethoprim Related Compound A 对照品/标准品1692505 甲氧苄啶 Trimethoprim 对照品/标准品1692006 盐酸曲美苄胺 Trimethobenzamide Hydrochloride 对照品/标准品1690000 三甲双酮 Trimethadione 对照品/标准品1689001 酒石酸阿利马嗪 Trimeprazine Tartrate 对照品/标准品1687006 盐酸苯海索 Trihexyphenidyl Hydrochloride 对照品/标准品1686310 曲氟尿苷杂质A Trifluridine Related Compound A 对照品/标准品1686309 曲氟尿苷 Trifluridine 对照品/标准品1686003 盐酸三氟丙嗪 Triflupromazine Hydrochloride 对照品/标准品1685500 2--chlorobenzophenone 对照品/标准品1685000 盐酸三氟拉嗪 Trifluoperazine Hydrochloride 对照品/标准品1683606 枸橼酸三乙酯 Triethyl Citrate 对照品/标准品1683504 盐酸曲恩汀 Trientine Hydrochloride 对照品/标准品1683005 曲地氯铵 Tridihexethyl Chloride 对照品/标准品1682217 三氯生杂质混合物A Triclosan Related Compounds Mixture A 对照品/标准品1682206 三氯生 Triclosan 对照品/标准品1681000 三氯噻嗪 Trichlormethiazide 对照品/标准品1680801 美曲膦酯 Trichlorfon 对照品/标准品1680685 三丁基氧化磷 Tributyl Phosphine Oxide 对照品/标准品1680608 枸橼酸三丁酯 Tributyl Citrate 对照品/标准品1680506 三唑仑CIV Triazolam CIV 对照品/标准品1680030 氨苯蝶啶杂质C Triamterene Related Compound C 对照品/标准品1680029 氨苯蝶啶杂质B Triamterene Related Compound B 对照品/标准品1680018 氨苯蝶啶杂质A Triamterene Related Compound A 对照品/标准品1680007 氨苯蝶啶 Triamterene 对照品/标准品1679008 己曲安奈德 Triamcinolone Hexacetonide 对照品/标准品1678005 醋酸曲安西龙 Triamcinolone Diacetate 对照品/标准品1677002 曲安奈德 Triamcinolone Acetonide 对照品/标准品1676000 曲安西龙 Triamcinolone 对照品/标准品1675007 三醋汀 Triacetin 对照品/标准品1674004 维 A 酸 Tretinoin 对照品/标准品1673839 醋酸群勃龙系统适用性实验用混合物 CIII Trenbolone Acetate System Suitability Mixture CIII 对照品/标准品1673828 醋酸群勃龙 CIII Trenbolone Acetate CIII 对照品/标准品1673806 群勃龙CIII Trenbolone CIII 对照品/标准品1673715 海藻糖 Trehalose 对照品/标准品1673500 盐酸曲唑酮 Trazodone Hydrochloride 对照品/标准品1673012 曲沃前列素杂质A Travoprost Related Compound A 对照品/标准品1673001 曲沃前列素 Travoprost 对照品/标准品1672916 苯环丙胺杂质A Tranylcypromine Related Compound A 对照品/标准品1672905 硫酸苯环丙胺 Tranylcypromine Sulfate 对照品/标准品1672803 反铂 Transplatin 对照品/标准品1672756 氨甲环酸杂质C Tranexamic Acid Related Compound C 对照品/标准品1672745 氨甲环酸 Tranexamic Acid 对照品/标准品1672712 群多普利杂质D Trandolapril Related Compound D 对照品/标准品1672701 群多普利杂质C Trandolapril Related Compound C 对照品/标准品1672687 群多普利 Trandolapril 对照品/标准品1672621 曲马多杂质B Tramadol Related Compound B 对照品/标准品1672610 曲马多杂质A Tramadol Related Compound A 对照品/标准品1672600 盐酸曲马多 Tramadol Hydrochloride 对照品/标准品1672337 托拉塞米杂质C Torsemide Related Compound C 对照品/标准品1672326 托拉塞米杂质B Torsemide Related Compound B 对照品/标准品1672315 托拉塞米杂质A Torsemide Related Compound A 对照品/标准品1672304 托拉塞米 Torsemide 对照品/标准品1672210 托吡酯杂质A Topiramate Related Compound A 对照品/标准品1672206 托吡酯;托佩马特 Topiramate 对照品/标准品1672100 含番茄红素的番茄提取物 Tomato Extract Containing Lycopene 对照品/标准品1672020 对甲苯磺酰胺 p-Toluenesulfonamide 对照品/标准品1672010 邻甲苯磺酰胺 o-Toluenesulfonamide 对照品/标准品1671006 托萘酯 Tolnaftate 对照品/标准品1670502 托美丁钠 Tolmetin Sodium 对照品/标准品1670229 托卡朋杂质 B Tolcapone Related Compound B 对照品/标准品1670218 托卡朋杂质 A Tolcapone Related Compound A 对照品/标准品1670207 托卡朋 Tolcapone 对照品/标准品1670003 甲苯磺丁脲 Tolbutamide 对照品/标准品1669004 盐酸妥拉唑林 Tolazoline Hydrochloride 对照品/标准品1668001 妥拉磺脲 Tolazamide 对照品/标准品1667938 替扎尼定杂质C Tizanidine Related Compound C 对照品/标准品1667924 替扎尼定杂质B Tizanidine Related Compound B 对照品/标准品1667916 替扎尼定杂质A Tizanidine







 我要推广仪器
我要推广仪器
 下载APP
下载APP