怎么分离亚氨基二乙酸和氨基三乙酸,样品中氨基三乙酸含量少
因工作需要,急需亚氨基二乙睛、亚氨基二乙酸化学法分析方法。我手里已有高效液相色谱分析方法,但单位没有液相色谱设备,只能干着急啊。
钴(Ⅲ)亚氨基二乙酸的配合物 怎么翻译啊?谢谢了
我要做氨基三乙酸和亚氨基二乙酸的分离,但是在C18柱上试了好几种方法都没有保留,请专家帮忙
用离子色谱测氨基酸一直测得挺好,中间有事停过一天的机,没开机,于是第二天继续测时,发现标准品中少了几个峰,而且乙酸钠的峰也没了。打电话咨询工程师,工程师说我的氢氧化钠浓度有问题,偏高,经过调试之前少了的那几个氨基酸又出来了,但是峰很小;另外,乙酸钠的峰也出来了,可是,又出现了问题,我同一个样品重复进样后乙酸钠的峰又没了。咨询工程师后,说色谱柱可能有问题,于是我又换了一个色谱柱,但是还是出现同样的问题,请高手帮个忙。
用离子色谱测氨基酸一直测得挺好,中间有事停过一天的机,没开机,于是第二天继续测时,发现标准品中少了几个峰,而且乙酸钠的峰也没了。打电话咨询工程师,工程师说我的氢氧化钠浓度有问题,偏高,经过调试之前少了的那几个氨基酸又出来了,但是峰很小;另外,乙酸钠的峰也出来了,可是,又出现了问题,我同一个样品重复进样后乙酸钠的峰又没了。咨询工程师后,说色谱柱可能有问题,于是我又换了一个色谱柱,但是还是出现同样的问题,请高手帮个忙。
ICP-AES一般是检测无机元素含量,主要是金属元素。我现有个疑问,亚胺基二乙酸想要检测一下其中的元素含量,并且它可以溶于水,那么水溶解以后是否就可以上机检测了?因曾做过这一类型的有机物,进样到ICP-AES中会熄火,不知道是不是因为有机物的原因,所以不太敢检测亚胺基二乙酸,检测有机物对仪器影响大吗?会不会把仪器烧了?请各位支招。谢谢!
请问哪里可以买到敌鼠钠盐标准品和氟乙酸标准品啊?
用戴安的离子色谱测水中二氯乙酸、三氯乙酸,到目前为止有可以溯源用的国标或是行业标准吗?如果还没有,CMA评审的时候非标方法确认很麻烦吗?有人做过这方面的工作吗,能不能借鉴一下。
卤乙酸的水溶液可以直接进HP-5色谱柱吗? 测定水中卤乙酸含量,可以用卤乙酸的标准品的水溶液做浓度梯度,绘制标准曲线吗? 问题补充: 使用的是ECD检测器。需要绘制卤乙酸的标准工作曲线。是卤乙酸用水溶,还是卤乙酸用甲醇做溶剂?能直接水溶卤乙酸进HP-5的色谱柱吗?
哪位好人有三氟乙酸、N-羟基琥珀酰亚胺、二环己基碳二亚胺的质量标准啊?请大家帮助。
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
[b]关于食品添加剂新品种氨基乙酸(羟基乙腈法)等的公告 2017年第3号 根据[b]《食品安全法》[/b]规定,审评机构组织专家对食品添加剂新品种氨基乙酸(羟基乙腈法)、食品用香料新品种乙基芳樟基醚和食品添加剂β-胡萝卜素扩大使用范围的安全性评估材料审查并通过。 特此公告。 附件:1. [url=http://file2.foodmate.net/wenku/20170321113512288.docx]食品添加剂新品种氨基乙酸(羟基乙腈法)[/url] 2. [url=http://file2.foodmate.net/wenku/20170321113830207.docx]食品用香料新品种乙基芳樟基醚[/url] 3. [url=http://file2.foodmate.net/wenku/20170321113946516.docx]食品添加剂β-胡萝卜素扩大使用范围[/url][/b][align=right] 国家卫生计生委[/align][align=right] [/align][align=right] 2017年3月8日[/align]
氨基酸标准品哪里买?
乙二醇双(2-氨基乙基醚)四乙酸的酸值怎么滴定?
最近要用高效液相PITC柱后衍生检测食品中氨基酸含量,我看很多文献都有氨基酸标准品,但是有的用了18种,也有用17种,15种的,就像问一下各位大佬,这个是有什么标准关于检测那一类食品一定要检测哪几种氨基酸嘛
大家好,我一直用安捷伦1120LC做17种氨基酸的含量测定,但是在检测过程发现因检测氨基酸的量很低,买的安捷伦的标准有时会偏低,使测定结果波动很大,想请问各位,买哪里的氨基酸标准比较稳定准确?再者,在检测氨基酸怎样才能提高准确度?
饮用水一般来源于自来水、桶装水和井水。自来水需经过消毒后才能饮用,其消毒方式一般包括氯消毒(液氯、次氯酸钠消毒等)和二氧化氯消毒。氯消毒因成本低廉的优点,目前是我国大型水厂的主流消毒方式。除卤代烃外,常见的含氯消毒副产物还有亚氯酸盐、氯酸盐、二氯乙酸和三氯乙酸等。这四种消毒副产物目前成为生活饮用水的常规检测项目,因此如何分离这四种消毒副产物成为目前一大热点。本文探索并开发[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]分离亚氯酸盐、氯酸盐、二氯乙酸和三氯乙酸的方法。首先是色谱柱和定量环的选择。由于一般饮用水中亚氯酸盐、氯酸盐、二氯乙酸和三氯乙酸在水中的检测值较低,因此需要采用大定量环测定,选择定量环体积为是500μL。实验所用 IonPac AS19分离柱亲水性好,柱容量高,能够满足生活饮用水中常见离子、卤乙酸及卤氧乙酸的同时测定。选择KOH作为淋洗液,利用淋洗液在线发生技术实现梯度洗脱,经过 AERS4 mm自动再生微膜抑制器抑制后产物为水,背景电导低,水负峰不明显,能够实现大体积进样,显著提高方法的灵敏度。淋洗液梯度的选择。选择以初始浓度分别为8、12、15 mmol/L来进行实验,结果表明,当初始浓度为8 mmol/L时,分离效果较好,使用初始浓度为12 mmol/L时,三氯乙酸受到硫酸盐的前延展性峰的干扰,分离效果不好;当浓度为15 mmol/L时,出峰速度较快,四种消毒副产物分离效果不好。由于三氯乙酸极性较大,需要采用梯度淋洗方法将进行洗脱。当选择(20-32)min匀速升至25 mmol/L,保持2 min,可以将保留时间较长的三氯乙酸尽快洗脱出来,且分离效果较好,减少检测所用时间,增加方法的实用性。梯度洗脱程序如表[align=center]表 四种消毒副产物的梯度洗脱程序[/align][table][tr][td]时间(min)[/td][td]梯度浓度C[sub]NaOH[/sub](mmol/L)[/td][/tr][tr][td]0-20[/td][td]8[/td][/tr][tr][td]20-32[/td][td]8-25[/td][/tr][tr][td]32-34[/td][td]25[/td][/tr][tr][td]34[/td][td]8[/td][/tr][/table]实际样品的测定。先对预先活化Ag柱、Ba柱和H柱,分别用注射器以2 mL/min 的流速将10mL超纯水过柱,静置10 min使其充分平衡。然后直接取适量水样,以2 mL / min的速度依次通过串联的Ag 柱、Ba 柱、H柱和0. 22 μm针式滤器,弃去前面 6 mL后开始收集滤液,滤液直接进样测试。可以明显去除氯离子和硫酸盐的含量,减少干扰峰的影响。实验中注意事项和建议先使用标准溶液分离这四种消毒副产物,再对三氯乙酸加标水样进行测定分离,确保三氯乙酸和硫酸盐可以有很好的分离度。二氧化碳装置使用。如果装有二氧化碳装置会大大降低硫酸根前延展性峰的干扰。使分离效果更好。
购买了17种混合氨基酸标准品,卖方没有给任何说明,怎么确定对应的氨基酸种类啊?很急,麻烦各位有经验的给我指出一条明路。谢谢了!
请问乙二胺四乙酸分析纯能直接配制EDTA标准溶液吗?请如果能配制有方法吗,比如配0.1mol/L的,请各位帮忙,我手里现在只有乙二胺四乙酸分析纯
我以前搞过段时间食品中氨基酸分析,以下是一点自己的笔记。供参考。由于涉及到了一些公司的内容。所以,我不敢透露更具体的。理解。主要样品:含脂肪的肉汤,肉块等。氨基酸分析测试室使用安捷伦1100系列HPLC仪器,通过柱前衍生法分析氨基酸。我司提供食品,保健品,饲料等游离和水解氨基酸含量测试。仪器配置和原理:四元泵梯度切换——自动进样器——柱——紫外检测器——记录仪自动进样器是通过吸取OPA(邻苯二醛)和FMOC-Cl加入样品和标准品中,使样品和标准品中的氨基酸接上荧光基团,在分析柱中分离,后用荧光检测器检测。荧光法检测精度高,检测限低。样品前处理原理和操做步骤:游离氨基酸前处理原理:通过使用三氯乙酸溶液作为蛋白质变性剂,沉淀蛋白质,而不破坏溶液中氨基酸。与公司前处理比较:公司使用热水浴提取固体或液体样品。该方法不能有效地去处样品中(汤,肉等)蛋白质,致使样品中蛋白质悬浮于样品中,污染色谱柱,仪器。由于样品处理简单,对于复杂样品可能提取效率稍差。大致操作步骤:称样——10%三氯乙酸定容——放置——滤过——离心方法优点:能有效去处蛋白质,而不破坏样品中氨基酸。水解氨基酸原理:分析蛋白质或多肽氨基酸组成,需将各肽键裂解为游离氨基酸。一般使用强酸或强碱。本次学习是使用6mol/L盐酸消解,提取。操作步骤:称样于消解管——加盐酸——抽真空——封口——烘箱消解——定容——蒸干——加水溶解——离心——上样分析
氨基酸是氨基和羧基的一类有机化合物的通称。生物功能大分子蛋白质的基本组成单位,是构成动物营养所需蛋白质的基本物质。是含有一个碱性氨基和一个酸性羧基的有机化合物。无色晶体,熔点极高,一般在200℃以上。不同的氨基酸其味不同,有的无味,有的味甜,有的味苦。各种氨基酸在水中的溶解度差别很大,并能溶解于稀酸或稀碱中,但不能溶于有机溶剂。近年来,随着技术的发展,检测氨基酸的种类及含量的方法很多,目前测定氨基酸的方法主要有氨基酸分析仪法、高效液相色谱法、毛细管电泳法及液相色谱-质谱联用法等。本文主要是通过高效液相串联质谱法来对16种氨基酸的含量进行测定,方法简单,快速,不用柱前衍生,节省试剂和成本,为保健品中氨基酸的含量测定提供了新方法。1实验部分1.1 仪器和试剂 16种氨基酸的混合标准品,(包括丙氨酸、精氨酸、天冬氨酸、胱氨酸、谷氨酸、组氨酸、亮氨酸、异亮氨酸、赖氨酸、蛋氨酸、苯丙氨酸、脯氨酸、苏氨酸、酪氨酸、缬氨酸、丝氨酸)安捷伦;乙腈(色谱纯)CNW;乙酸铵(色谱纯);乙酸(色谱纯)。超声波清洗器(EQ-500);DHG-9240A型电热恒温鼓风干燥箱(上海鸿都电子科技有限公司);BPZ-6090Lc型真空干燥箱(上海一恒科学仪器有限公司);高效液相色谱仪(安捷伦1260);色谱柱:安捷伦poroshell 120 EC-C18柱(4.6mm×50mm,2.7μm);质谱(API3200)美国AB公司。1.2 色谱条件色谱柱:以十八烷基硅烷健合硅胶为填充剂(柱长15cm,内径4.6mm,粒径2.7μm )或同等性能的色谱柱;柱温:40℃;流速:0.5 ml/min;进样量:5μL ;流动相:流动相A:称取0.386g乙酸铵,加水500mL溶解,用乙酸调pH至3.5;流动相B:乙腈,梯度洗脱:时间(分钟)流动相A(%)流动相B(%)0.0090100.00-5.0090→8010→205.00-9.0080→9020→101.3质谱条件1.3.1以电喷雾电离源(ESI)阳离子模式,气帘气20psi,碰撞气6,喷雾电压5500V,离子源温度600°C,雾化气是50 psi,辅助气是50 psi,时间9min。1.3.2 16种氨基酸的质谱参数,见表1。表1 16氨基酸的质谱参数氨基酸Q1Q3TIMEDPEPCECXP丙氨酸90.144.2353510153精氨酸175.2116353010153天冬氨酸134.474354110153胱氨酸241.174.11002610153谷氨酸148.2102353010153组氨酸156.1110353510163亮氨酸132.386.1352610113异亮氨酸132.286352610113赖氨酸147.3130353010153蛋氨酸150.1104352510103苯丙氨酸166.4120352510143脯氨酸116.270353510143苏氨酸120.174.3354110153酪氨酸182.3136353510153缬氨酸118.272354110153丝氨酸106.160.23530101331.4 标准品溶液的配制量取16种氨基酸的混合标准品1ml,置于50ml容量瓶,加0.1%甲酸水定容,摇匀即得标准品贮备液。1.5 供试品溶液的制备 精密称取样品约0.1g,置于25ml磨口的具塞比色管内,加6mol/L盐酸15ml,加入0.2g苯酚,用旋转混合仪和超声仪使样品充分分散并溶解,充氮气,盖紧塞子,置于110℃±1℃的恒温干燥箱内,水解22小时,取出冷却,过滤,用纯化水冲洗比色管,将水解液全部转移至50mL容量瓶中,用纯化水定容至刻度,摇匀,精密吸取1mL于10mL容量瓶中,置于真空干燥箱内,于40℃~50℃减压干燥(真空干燥箱内放入五氧化二磷作为干燥剂),干燥后残留物用0.1%甲酸水定容至刻度,摇匀,经0.45μm的微孔滤膜过滤,即得。1.6 线性实验将标准品贮备液稀释成0.004nmol/μl,0.008nmol/μl,0.012nmol/μl,0.016nmol/μl,0.020nmol/μl浓度梯度,每个浓度以5μL注入色谱系统。1.7 精密度实验将标准品贮备液稀释至一定的浓度,按色谱、质谱条件进行测定,连续进样6次。1.8 加样回收率实验精密称取五分的供试品,每份加入0.020nmol/μl标准品溶液5ml,定容。以5μL注入色谱系统。1.9供试品的测定 把1.5制备好的供试品溶液,以5μL注入色谱系统。以标准液的峰面积和浓度,通过外标法做曲线算出样品的浓度。2 结果与讨论2.1 色谱行为由于保健品中的氨基酸,用高效液相荧光法要进行柱前衍生,成本很高而采用液质联用法,不需要衍生,可大大节约成本,用流动相(0.35g乙酸铵加水0.5L溶解,然后加入0.5L乙腈,混合,用乙酸调pH=3.5)时,5min就能把16种氨基酸全部流出。采用液质联用法,通过MRM模式进行母离子及相应的子离子进行检测,能达到准确的分离和定量,见图1 。 从左到右,分别为丙氨酸、精氨酸、天冬氨酸、胱氨酸、谷氨酸、组氨酸、亮氨酸、异亮氨酸、赖氨酸、蛋氨酸、苯丙氨酸、脯氨酸、苏氨酸、酪氨酸、缬氨酸、丝氨酸。图1 16种氨基酸标准品的碎片离子色谱峰2.2 质谱行为在ESI(+)阳离子检测方式下,16种氨基酸的质谱定量分析,在质谱条件下,失去NH3形成+ 峰,或失去HCOOH重排生成+ 峰,从而进行色谱分离。2.3 线性回归方程和最低检测限以标准品的5个梯度浓度与其峰面积进行线性回归,拟合线性回归方程为Y=aX + b,相关系数r2 及最低检测限(S/N(信噪比)=3时,可测得标准品的最低检测限)见表2。表2 16种氨基酸线性回归方程、相关系数、最低检测限氨基酸Y=aX + b相关系数(r2)最低检测限(nmol/μl)丙氨酸Y=3.09e+006X + 6.95e+0030.99810.0013精氨酸Y=2.31e+007X + 1180.99990.0005天冬氨酸Y=3.45e+006X + 1.07e+0040.99760.0008胱氨酸Y=8.44e+005X + 1.72e+0030.99660.0006谷氨酸Y=7.6e+006X + 1.46e+0040.99840.0006组氨酸Y=7.58e+007X + 5.98e+0030.99990.0004亮氨酸Y=6.06e+007X + 5.24e+0040.99880.0001异亮氨酸Y=8.15e+007X + 6.26e+0040.99960.0001赖氨酸Y=1.24e+007X + 2.67e+0040.99780.0003蛋氨酸Y=1.34e+007X + 5.14e+0030.99890.0001苯丙氨酸Y=7.42e+007X + 4.08e+0040.99910.0001脯氨酸Y=7.9e+007X + 5.57e+0040.99970.0003苏氨酸Y=9.67e+006X + 2.49e+0040.99770.0003酪氨酸Y=1.76e+007X + 2.89e+0040.99920.0003缬氨酸Y=2.41e+007X + 3.31e+0040.99740.0003丝氨酸Y=1.06e+007X + 2.63e+0040.99810.00132.4 方法的精密度和重复性 取浓度0.012nmol/μl的16种氨基酸混合标准品连续进样6次。计算峰面积和保留时间的RSD值。结果见表3。表3 16种氨基酸峰面积和保留时间的相对标准偏差氨基酸峰面积RSD(%)保留时间RSD(%)丙氨酸4.480.57精氨酸4.670.34天冬氨酸4.570.44胱氨酸3.780.56谷氨酸3.360.24组氨酸3.330.13亮氨酸2.750.1异亮氨酸2.930.23赖氨酸4.790.43蛋氨酸3.920.09苯丙氨酸2.170.11脯氨酸4.540.28苏氨酸4.110.37酪氨酸3.610.13缬氨酸2.350.23丝氨酸3.280.162.5加样回收率实验,见表4。表4 16种氨基酸峰的回收率实验结果氨基酸平均回收率(%)RSD(%)丙氨酸94.114.05精氨酸82.912.73天冬氨酸82.694.66胱氨酸83.153.95谷氨酸98.234.1组氨酸78.671.61亮氨酸98.284.99异亮氨酸102.713.03赖氨酸98.584.49蛋氨酸95.983.34
有谁能告诉我维生素A乙酸酯标准品99%的都买的哪里的?
本标准规定了食品添加剂乙二胺四乙酸铁钠的技术要求、试验方法、检验规则、标志、包装、运输和贮存及保质期。
(1)原理试样经处理后,在pH6左右的溶液中,镉离子与二硫腙形成配合物,并经乙酸丁酯萃取分离,导入原子吸收仪中,原子化以后,吸收228.8nm共振线,其吸收值与镉含量成正比,与标准系列比较定量。(2)试剂 氨水、混合酸、1g/L二硫腙-乙酸丁酯溶液(称取0.1g二硫腙,加10mL三氯甲烷溶解后,再加乙酸丁酯稀释至100rnL,临用时配制)、2mo1/L柠檬酸钠缓冲液(称取226.3g柠檬酸钠及48.46g柠檬酸,加水溶解,必要时加温助溶,冷却后加水稀释至500mL,临用前用1g/L二硫腙-乙酸丁酯溶液处理以降低空白值)、镉标准储备溶液和标准使用液的配制与碘化钾-4-甲基戊酮-2法中的相同。(3)仪器原子吸收分光光度计。(4)分析步骤①试样处理对于谷类要去除其中的杂物及尘土,必要时除去外壳。对于豆类,取可食部分洗净晾干,切碎充分混匀。②样品消化称取5.00g上述试样,置于250mL高型烧杯中,加15mL混合酸,盖上表面皿,放置过夜,再于电热板或电砂浴上加热。消化过程中,注意勿使干涸,必要时可加少量硝酸,直至溶液澄明无色或微带黄色。冷后加25mL水煮沸,除去残余的硝酸至产生大量白烟为止,如此处理两次,放冷。以25mL水分数次将烧杯内容物洗入125mL分液漏斗中。取与处理样品相同量的混合酸、硝酸按同一操作方法做试剂空白试验。③萃取分离 吸取0、0.25mL、0.50mL、1.50mL、2.50mL、3.50mL、5.0mL镉标准使用液(相当于0、0.05μg、0.1μg、0.3μg、0.5μg、0.7μg、1.0μg镉)。分别置于125mL分液漏斗中,各加盐酸(1+11)至25mL。向试样品处理溶液、试剂空白液及镉标准溶液各分液漏斗中各加5mL柠檬酸钠缓冲液(2mol/L),以氨水调节pH至5~6.4,然后各加水至50mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),以氨水调节pH至5~6.4,然后各加水至501mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),振摇2min,静置分层,弃去下层水相,将有机层放入具塞试管中,备用。④测定测定方法与碘化钾-4-甲基戊酮-2法中的相同。⑤结果计算 样品中镉的含量按下式进行计算。X=/(m×1000)式中,X为试样中镉的含量,mg/kg;A1为测定用试样液中镉的质量,μg;A2为试剂空白液中镉的质量,μg;m为试样质量或体积,g或mL。计算结果保留两位有效数字。⑥精密度 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%。
这几天用岛津的碳18分析氨基酸,用的异硫氰酸苯酯为衍生剂。 用水代替氨基酸标准品,会出现很多杂峰,所以想应该是衍生过程引进的。衍生的方法是 标准品+三乙胺乙腈溶液+异硫氰酸苯酯乙腈溶液 反应一个小时候用正己烷萃取各位大虾有更好的衍生方法么?本实验室没有氮吹仪,没法吹干哦
氨基乙酸 掩蔽剂各位大虾!氨基乙酸常用来掩蔽那些金属离子呢?我想掩蔽镍和钴的混合溶液中得镍离子不知可行不?有木有其他好的掩蔽剂可以掩蔽钴中得镍?
关于对《黄酒中氨基酸的测定方法》行业标准(征求意见稿)征求意见的通知 全国酿酒标准化技术委员会委员、各有关单位、专家: 根据发改办工业1242号文件下达的行业标准计划任务,《黄酒中氨基酸的测定方法》列入行标计划,由全国食品发酵标准化中心归口管理。 全国酿酒标准化技术委员会秘书处按照标准制修订工作程序,组织完成了《黄酒中氨基酸测定方法》行业标准征求意见稿,现向有关行业部门、协会以及相关生产、销售、科研、检测和用户等单位广泛征求意见。通知如下:1.相关附件可以从http://www.cnif.cn/上下载。2.按照附表格式填写意见反馈表,若没有意见也请返回。3.请于2011年3月25日前反馈至秘书处。秘书处联系人: 钟其顶 郭新光 电话:010-64647778,64666533 传 真: 010-64677607 邮箱: TC471@scff.org.cn附: 1.《黄酒中氨基酸的测定方法》行业标准(征求意见稿)2.《黄酒中氨基酸的测定方法》行业标准编制说明(征求意见稿)3.《黄酒中氨基酸的测定方法》行业标准(征求意见稿)意见反馈表 全国酿酒标准化技术委员会秘书处 二O一一年二月二十四日 相关附件下载
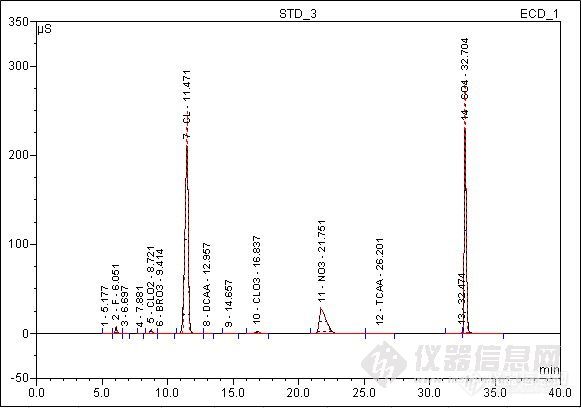
离子色谱法测定水中二氯乙酸和三氯乙酸检测实施细则1 目的规范离子色谱测定生活饮用水和水源水中二氯乙酸和三氯乙酸检测方法,保证检测工作顺利进行。2 方法的适用范围适用于使用戴安ICS-900离子色谱仪对生活饮用水和水源水中二氯乙酸和三氯乙酸进行检测。该方法对这两种消毒副产物的方法检出限(MDL)分别为:二氯乙酸:1.5μg/L;三氯乙酸:1.8μg/L,定量检测限(LOQ)分别为:二氯乙酸:6.0μg/L三氯乙酸:7.2μg/L。3 仪器3.1 ICS-900离子色谱仪:AS19分析柱、AG19保护柱、ASRS300抑制器、ED50电导检测器3.2 AS-DV自动进样器3.3 CR-ATC淋洗液自动生成器4 试剂4.1 纯水:含各种待测阴离子应低于仪器的最低检出限,使用前应测定空白值。4.2 二氯乙酸和三氯乙酸标准物质,由上海安谱科学仪器有限公司提供。二氯乙酸标准品:1g;三氯乙酸标准品:1g(纯度为99.40%)。5 水样5.1 水样的采集及储存:水样采集在聚乙烯瓶中,于4℃冰箱内保存。5.2 水样的预处理:经0.2μm孔径的微孔滤膜过滤后可直接进样;若样品中碳酸根的质量浓度在200 mg/L以上,则要经过酸化除去大量存在的碳酸根,然后将样品在真空装置中进行脱气,再进样分析。样品测定时应注意的问题样品中某些组分浓度太高,应选择适当的稀释倍数稀释样品后测定。如果已检测了高组分的样品,并对下一样品的测定产生了影响,建议进1一2针去离子水测定,将高组份完全洗脱后再测下一个样品。由于进样量小,操作装应严格防止纯水和器皿在水样预处理过程中的污染,以确保分析的准确性。6 试验6.1 色谱条件进样体积:300μL;流量:1.0mL/min。表5.1.1.1 淋洗液梯度程序时间/(min)氢氧化钾浓度/(mmol/L)0.010.025.0
[font=SimSun, STSong, &]我的食品生产许可证上的执行标准是SB/T10439-2007产品分类是酱腌菜产品明细是盐水食用菌,请问的我生产的盐水食用菌允许使用乙二胺四乙酸二钠吗?2760上边按照酱腌菜的标准是允许的,按食用菌制品的标准是不允许使用的,我是应该按照食用菌制品的标准不允许使用呢还是应该按照生产许可证上的酱腌菜标准允许使用呢?有没有专业人士帮忙解答啊?[/font]







 我要推广仪器
我要推广仪器
 下载APP
下载APP