用盐酸克伦特罗配制克伦特罗标准溶液,是否要出去盐酸根离子称取标准品质量。
我们参与能力验证用[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定金属元素Cu和Pb,Cu样品介质为盐酸5%溶液,Pb样品介质为硝酸5%溶液,那么配制标准系列时Cu和Pb也分别是5%盐酸和5%硝酸?平时我们一般是0.2%硝酸.总之标准系列的酸度要和样品一致吗?
哪位大侠知道哪里有卖孔雀石绿的盐酸盐标准品的?谢谢![em23] [em27]
我需要D5-莱克多巴胺标准品,发现只有D5-莱克多巴胺盐酸盐标准品,测食品中残留量是不是一样的用
看到饲料中盐酸克伦特罗的检测,与大家分享! 饲料中盐酸克伦特罗的检测1.盐酸克伦特罗准确定性定量产品配置采用SPE-LC对饲料中盐酸克伦特罗准确定性定量,参考配置如下: 仪器配置:P1201等度系统基本配置 色谱柱:Hypersil BDS C18色谱柱(4.6×250mm,5µm) 固相萃取柱: Thermo HyperSep Retain CX(3ml,200mg)参考前处理设备 超声波清洗器 离心机 氮吹仪 酸度调节计2.样品处理准确称取饲料5g左右,准确加入0.5%偏磷酸50mL,超声提取15min,每5min取出振摇一次,超声结束后手摇10s,并取上清液以4000r/min离心10min,取上清液10mL置分液漏斗中滴加氢氧化钠溶液,充分振摇,调pH值至12左右,溶液用3-mL乙醚萃取两次,萃取液用无水硫酸钠干燥,于50℃下干燥,残渣用0.01mol/L盐酸溶液2mL溶解,待净化。3.样品净化依次用3mL甲醇、3mL超纯水和3mL 0.5mol/L高氯酸润洗固相萃取小柱,取待净化液注入SPE小柱中,依次用3mL超纯水、3mL甲醇淋洗,用3mL 5%的氨化甲醇进行洗脱,收集液用氮气吹干,残渣用40:60的甲醇和0.05%磷酸混合溶液溶解,过滤,即可。4.样品检测将净化的样品溶液注入液相色谱仪,在243nm下检测,谱图见图1。
酸度(盐酸或硝酸超过50%对火焰测守有影响吗
现在很多目标物买的标准品都是对应的盐酸盐,除了标品制备更稳定外,还出于什么目的?在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]中目标物和对应的盐酸盐为何保留时间无区别,另外如果在质谱中,盐酸盐的标品和目标物是否会有区别
食品安全国家标准 食品营养强化剂 L-盐酸赖氨酸
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
项目:含量测定(3.2.P.5.2.7)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:色谱柱(柱长:150mm,内径:4.6mm,填料:C18,填料粒径:5μm)http://ng1.17img.cn/bbsfiles/images/2013/01/201301081630_419121_1621890_3.gifUV检测器(检测波长:225nm)柱温:30℃流动相:0.2mol/L磷酸二氢钾-0.2mol/L磷酸-乙腈(7:7:5)流速:1.0ml/min运行时间:约25分钟系统适用性:理论板数按盐酸坦洛新峰计算不低于2000。具体试验操作:取本品20片,精密称定,研细,精密称取适量(约相当于盐酸坦洛新0.2mg),置50ml量瓶中,加流动相适量振摇后超声使溶解,放冷至室温,加流动相稀释至刻度,摇匀,滤过,精密量取续滤液20μl注入液相色谱仪中,记录色谱图;另精密称取盐酸坦洛新对照品2mg置50ml量瓶中,加流动相振摇后超声使溶解并稀释至刻度,摇匀,滤过,精密量取续滤液1ml置10ml量瓶中,加流动相稀释至刻度,作为对照品溶液,精密量取对照品溶液20μl注入液相色谱仪中,记录色谱图。按外标法以峰面积计算,即得。计算公式:http://ng1.17img.cn/bbsfiles/images/2013/01/201301081634_419123_1621890_3.gif标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。3.2.P.5.3.5含量测定含量测定方法学验证结果概要项目验证结果波长选择225nm。流动相选择0.2mol/L磷酸二氢钾-0.2mol/L磷酸-乙腈(7:7:5)。进样精密度试验连续测量6次,主峰保留时间和峰面积RSD均小于2.0%。线性关系试验浓度在2460ng/ml~5740ng/ml,直线方程为y=107.8x﹢5880.1,R2=0.9992,相关系数r=0.9996,截距为5880,小于100%浓度溶液主峰面积的2.0%(8841)。溶液稳定性试验室温条件下置12个小时,测得峰面积的RSD为1.3%回收率试验平均回收率为100.3%,在98.0%~102.0%范围内,RSD为1.0%(n=9),小于2.0%。重复性试验试验结果在95.3%~100.5,RSD值为2.0%。耐用性同有关物质项含量测定结果上市品和自制品均符合规定3.2.P.5.3.5.1波长选择参照盐酸坦洛新缓释片药品质量标准YBH19552005含量测定项、盐酸坦洛新原料药质量标准YBH03262005有关物质项和本品有关物质研究试验结果,检测波长选定为225nm。3.2.P.5.3.5.2[
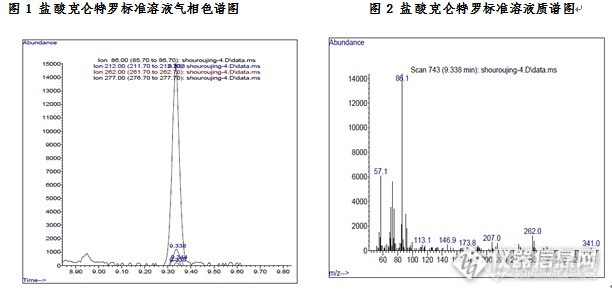
动物源性食品中盐酸克仑特罗检测的固相萃取方法(Silibase™ C8/SCX)一、实验目的本研究利用固相萃取法作为猪肉样品的前处理方法,GC-MS法作为检测手段。该方法可简化猪肉样品的前处理过程,节省有机溶剂的使用,操作简便。 二、实验目标物盐酸克仑特罗(CAS;21898-19-1)三、应用范围本方法适用于动物源性食品中盐酸克仑特罗的GC-MS检测及确证。 四、参考标准农业部推行标准《NY/T 468-2006 动物组织中盐酸克伦特罗的测定气相色谱-质谱法》五、实验材料 C8/SCX固相萃取柱6mL/500mg。六、实验方法1、样品提取 称取均质试样5.0g试样(精确至0.01g)于50mL离心管中,加入15mL乙酸乙酯,再加入3mL10.0%碳酸钠溶液,然后以10000r/min以上的速度均质60s。盖上盖子以5000r/min的速度离心2min,吸取上层有机试剂于离心管中,在残渣中再加入10mL乙酸乙酯在旋涡混合器上混合1min,离心后吸取有机溶剂并合并提取液。在收集的有机试剂中加入5mL 0.1mol/L的盐酸溶液,涡旋混合30s。以5000r/min的速度离心2min,吸取下层溶液,同样步骤重复萃取一次,合并两次萃取液,用2.5mol/L氢氧化钠溶液调节pH至5.2。 2、SPE柱净化(1)活化:向C8/MCX复合柱小柱中依次加入5mL甲醇、水5mL和5mL 30mmol/L盐酸润洗固相萃取小柱。(2)上样和洗脱:将上述备用液过柱,依次用5mL水、5mL甲醇淋洗,真空抽干,用4%氨化甲醇5mL洗脱小柱,收集洗脱液。(3)衍生化及检测:0℃缓慢氮气流条件下吹至近干,加入甲苯100μL和双三甲基硅基三氟乙酰胺100μL,涡旋震荡20s,密封玻璃塞,置于80℃恒温烘箱中加热1h,冷却后加300μL甲苯,作为试样溶液,供气相色谱-质谱分析,上气相色谱-质谱仪测定。3、GC-MS条件气相色谱-质谱仪;色谱柱:HP-5MS进样口:220℃柱温:70℃(保持0.6min),以25℃/min升温至200℃(保持6min),以25℃/min升温至280℃(保持5min)载气:高纯He,流速:0.9mL/min进样体积:1μlGC/MS传输线温度:280℃溶剂延迟:8min分析器温度:230℃四级杆温度:150℃ 七、实验结果1、添加回收结果实验结果表明,C8/SCX复合固相萃取柱适用于动物组织中盐酸克伦特罗的预处理,能净化动物组织样品,实验加标回收率及RSD能满足定量实验的要求。表1动物源性食品中盐酸克伦特罗的添加回收结果 1 2 3 4 5 平均回收率(%) RSD(%) n=5 回收率(%) 85.6 92.4 94.1 95.7 87.6 90.8 4.23 2、标准溶液色谱图在GC-MS操作条件下,得到标准溶液色谱图如图1和图2http://ng1.17img.cn/bbsfiles/images/2015/08/201508141658_560811_3310_3.jpg
自己配制标准溶液,盐酸盐形态的克伦特罗,草酸盐形态的孔雀石绿标准溶液,称量时应称取多少g?目标称量量是50mg。
原子荧光测镉时使用液稀释问题镉储备液用盐酸稀释会有很大影响吗?镉在高盐酸酸度下会不会形成不易生成氢化物的化合物那?镉在什么介质中即稳定有益与原子荧光的发生?
一 酸度调节剂Acidity regulator酸度调节剂,或PH值控制剂,是用来调整或保持PH值(酸或碱)的一种食品添加剂。酸度调节剂可以是有机酸或无机酸、碱、中和剂或缓冲剂。酸度调节剂由其E编码标识,如E260(乙酸),或列在“食品酸度剂”下。经常使用的酸度调节剂是柠檬酸,乙酸和乳酸。碳酸钠、碳酸钾可用于面制食品中,盐酸、氢氧化钠属于强酸、强碱性物质,其对人体具有腐蚀性,只能用作加工助剂,要在食品完成加工前予以中和。简介 酸度调节剂亦称pH调节剂,是用以维持或改变食品酸碱度的物质。它主要用以控制食品所有需的酸化剂、碱剂以及具有缓冲作用的盐类。 酸化剂具有增进食品质量的许多功能特性,例如改变和维持食品的酸度并改善其风味;增进抗氧化作用,防止食品酸败;与重金属离子络合,具有阻止氧化或褐变反应、稳定颜色、降低浊度、增强胶凝特性等作用。酸化剂均有—定的抗微生物作用,尽管单独用酸来抑菌,防腐所需浓度太大,影响食品感官特性,难以实际应用,但是当以足够的浓度,选用一定的酸化剂与其他保藏方法如冷藏、加热等并用,可以有效地延长食品的保存期。至于对不同酸的选择、取决于酸的性质及其成本等。 酸味的刺激阈值用pH值来表示,无机酸的酸味阈值在3.4~3.5左右,有机酸的酸味阈值在3.7~4.9之间。大多数食品的pH值在 5~6.5之间,虽为酸性,但并无酸味感觉,若pH值在3.0以下,则酸味感强,难以适口。 酸度调节剂其有效应用主要受食品所需特性控制,通常以有机酸及具有缓冲作用的盐为主。又由于很多有机酸都是食品的正常成分,或参与人体正常代谢,因而安全性高,使用广泛。我国批准许可使用的酸度调节剂品种不少。但是,与国外许可使用的同类品种相比尚有一定差距。主要是缺少各种有机酸的盐。不过,当前重要的是加强应用开发,应尽量利用现有品种,针对不同食品原料,研制出具有各自不同风味特点而受人欢迎的食品。基本介绍 酸度调节剂为增强食品中酸味和调整食品中pH或具有缓冲作用的酸、碱、 盐类物质总称。规定允许使用的酸度调节剂有柠檬酸、柠檬酸钾、乳酸、酒石酸等17种,其中柠檬酸为广泛应用的一种酸味剂。柠檬酸、乳酸、酒石酸、苹果酸、柠檬酸钠、柠檬酸钾等均可按正常需要用于各类食品。 酸度调节剂亦称pH调节剂,是用以维持或改变食品酸碱度的物质。它主要有用以控制食品所需的酸化剂、碱剂以及具有缓冲作用的盐类。酸化剂具有增进食品质量的许多功能特性,例如改变和维持食品的酸度并改善其风味;增进抗氧化作用,防止食品酸败;与重金属离子络合,具有阻止氧化或褪变反应、稳定颜色、降低浊度、增强胶凝特性等作用。酸均有一定的抗微生物作用,尽管单独用酸来抑菌,防腐所需浓度太大,影响食品感官特性,难以实际应用,但是当以足够的浓度,选用一定的酸化剂与其他保藏方法如冷藏、加热等并用,可以有效地延长食品的保存期。至于对不同酸的选择,取决于酸的性质及其成本等。 酸度调节剂除可使用的酸度调节剂品种不少。但是,与国外许可使用的同类品种相比尚有一定差距,主要是缺少各种有机酸的盐。不过,当前重要的是加强应用开发,应尽量利用现有品种,针对不同食品原料,研制出具有各自不同风味特点,受人欢迎的加工食品。酸度调节剂除可调节食品的pH、控制酸度、改善风味之外,尚有许多其他功能特性。其有效应用主要受食品所需特性控制,通常以有机酸及具有缓冲作用的盐为主。又由于很多有机酸都是食品的正常成分,或参与人体正常代谢,因而安全性高,使用广泛。性质介绍 又称pH值调节剂。是用以维持或改变食品酸碱度的物质。主要有用以控制食品所需的酸化剂、碱剂以及具有缓冲作用的盐类。酸化剂具有增进食品质量的特性,如改变和维持食品的酸度并改善其风味;增进抗氧化作用,防止食品酸败;与重金属离子络合,具有阻止氧化或褐变反应、稳定颜色、降低浊度、增强胶凝特性等作用。酸均有一定的抗微生物作用,选用一定的酸化剂与其他保藏方法如冷藏、加热等并用,可以有效地延长食品的保质期。种类介绍 已批准许可使用的酸度调节剂有: 卫生标准 酸度调节剂 范围 本标准规定了食品添加剂的品种,使用范围及使用量。 酸度调节剂原理本标准适用于1995年、1996年度申请单位申报的新增食品添加剂的使用卫生标准以及某些现有品种的扩大使用范围。 标准为GB2760—1996《食品添加剂使用卫生标准》的续篇。 引用标准 下列标准所包含的条文,通过在本标准中引用而构成为本标准的条文。本标准出版时,所示版本均有效。所有标准都会被修订,使用本标准的各方应探讨使用下列标准最新版本的可能性。 GB2760一1996食品添加剂使用卫生标准 GBl2493—1990食品添加剂分类和代码 GBl4880—1994食品营养强化剂使用卫生标准 编辑本段实际运用 南京国海生物工程有限公司作为全国最大的DL-苹果酸制造商,市场份额占66%,产品技术指标达到甚至部分指标优于国际相关标准规定,但由于种种原因,在去年国家标准化管理委员会公布的国家标准立项计划中,《食品添加剂DL-苹果酸》国家标准话语权“花落旁家”,国海生物公司无缘参与。标准话语权的丧失将对国海生物公司的技术研发、生产控制、成品质量和销售等造成严重影响,很大程度制约着企业发展。得知消息后,标准化处与六合分局第一时间深入企业分析原因、查找问题,多次与全国有机化学标准化技术委员会协调沟通,并率企业赴北京向国家标准委和行业主管部门汇报情况、积极磋商,取得了国标委和行业的了解和支持,国海生物公司被批准为《食品添加剂DL-苹果酸》国家标准主导研制单位。6月19日,该国家标准行业预审会在六合成功举行,标志着我市又一重点项目关键技术夺回标准话语权。 随着《食品添加剂DL-苹果酸》国家标准的研制成功,DL-苹果酸作为食品添加剂中的一种酸度调节
[em0815] 哪为老师有盐酸多西环素和多西环素一水物标准BP2008和EP6.0版部分.请分享一下.不胜感激.急需.
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测环丙沙星,但标准品买的是盐酸环丙沙星,在配置时需要扣掉盐酸的质量吗?另外我用乙腈溶解发现溶解后的溶液白色浑浊,很多白色的悬浮物,加100ul甲酸后仍然浑浊,老师们有遇到过这种情况吗?
大家好, 有人能帮助提供我原料药品心得安(盐酸普萘洛尔)的中控分析方法吗?恳请大家帮助。
用HPLC测盐酸克伦特罗,按国标方法用水和甲醇做流动相,244nm做检测波长,发现标准品的峰拖尾特别严重,而且信号响应也特别低,发现可能式因为克伦特罗呈碱性,所用的又是C18柱,于是加入1%磷酸(PH=2.3),避免因为c18上的-H作用,可是发现效果也不好,拖尾照样拖,换了三根柱子都是这样的情况,很是郁闷,而且发现信号很弱很弱(在230nm时还稍微好点),这样拖尾的峰型和信号强度根本没有办法去定量。 希望各位指点,谢谢!
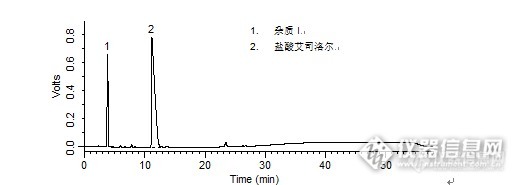
1. 杂质I2. 盐酸艾司洛尔 盐酸艾司洛尔样品制备 制备方法有关物质衍生溶液:取盐酸艾司洛尔对照品约10 mg,置10 mL量瓶中,加入1 mol/L盐酸溶液1 mL,放置30分钟,加1 mol/L的氢氧化钠溶液1 mL使中和,用流动相A 稀释至刻度,摇匀。分析条件 色谱柱Diamonsil C18(2) 250 x 4.6 mm,5 μm (Cat#:99603)流动相流动相A:乙腈:甲醇:磷酸盐缓冲液(取磷酸二氢钾3.0 g,加水至650 mL)=15:20:65流动相B:甲醇梯度流速1 mL/min柱温30 ℃检测器UV 222 nm进样量20 μL 色谱图有关物质衍生溶液http://ng1.17img.cn/bbsfiles/images/2016/04/201604211737_591070_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 分离度 1 3.842 6280189 655879 2747.059 0.670 -- 2 11.157 29271705 784686 1512.532 5.026 10.154 本品种同时使用了SpursilC18色谱柱,在药典规定条件下进行检测,满足药典要求。
我做火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url],发现有关技术人员有两种配制校准曲线的方法:1,100ML容量瓶先加水,再统一加1+1硝酸或盐酸2ML,最后加用不同体积的1%酸稀释好的标准溶液,配制校准曲线。2。100ML容量瓶先加用1%酸稀释好的不同体积的标准溶液,最后用1%酸定容。我当然知道第二个方法是国标方法,标准曲线的酸度保持一致,好。可是配大量的酸需要很大的体积,不方便。可是用第一个方法标准曲线的酸度不一致,如先加1+1硝酸或盐酸2ML,在加10ML标准溶液,用纯水定容后实际酸度是1.1%.不知影响试验结果吗?影响大吗?标准贮备液的中间液可以用纯水稀释吗?这样就避免了第一个方法的酸度不一致。但我不知用纯水稀释行吗?第二个问题:我看别人研究盐酸和硝酸对[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]吸光度的影响。难道在同浓度的盐酸和硝酸条件下,吸光度的数据是一样的吗?怎么我的老师说差别很大呢。而且铅在盐酸中应该沉淀啊?怎么火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]可以用盐酸介质啊?别人数据如下:无机酸的影响V/mL246810Co吸光度HCl0.1100.1040.1080.1130.117HNO30.0980.1070.1090.1130.119Ca吸光度HCl0.1650.1600.1690.1690.168HNO30.1600.1630.1600.1670.165Fe吸光度HCl0.1200.1190.1210.1200.126HNO30.1270.1200.1200.1210.123Pb吸光度HCl0.1030.1000.1010.1050.107HNO30.1020.1010.1050.1030.108结果表明在HNO3和盐酸体积分数2%~10%的酸度范围内对被测元素无影响,第三个问题,我用火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测镍,用1%硝酸定容,怎么数据特别不好,标准偏差很大?而且同用1%盐酸作介质的结果差别很大,不知为什么?相同样品同倍数稀释,一个1.6PPM。一个2.5PPM第四个问题,Cu,Zn,Pb,Cd,Fe,Mn,Ni,可以在一起配混标吗?最近焦头烂额,实在太忙了,试验连续做不完,没时间研究这些问题?我恭谨的等各位研究过这些问题的大侠和老师给我解惑?谢谢了
论坛里做过猪尿和猪肉中盐酸克仑特罗的朋友,你们做的回收率是多少?是采用哪个标准做的?我现在采用的是DB33/T623-2006和DB33/T624-2006这两个标准做的,回收率老是做不好
各位老师,关于71-3中0.07mol/L 的盐酸标定,有国标标准时说关于盐酸标定的么?
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7651724918 唑吡坦杂质A CIV Zolpidem Related Compound A CIV 对照品/标准品1724907 酒石酸唑吡坦 CIV Zolpidem Tartrate CIV 对照品/标准品1724893 唑吡坦 CIV Zolpidem CIV 对照品/标准品1724805 盐酸唑拉西泮 Zolazepam Hydrochloride 对照品/标准品1724769 硫酸锌 Zinc Sulfate 对照品/标准品1724747 氧化锌 Zinc Oxide 对照品/标准品1724689 齐留通杂质C Zileuton Related Compound C 对照品/标准品1724678 齐留通杂质B Zileuton Related Compound B 对照品/标准品1724667 齐留通杂质 A Zileuton Related Compound A 对照品/标准品1724656 齐留通 Zileuton 对照品/标准品1724532 齐多夫定杂质C(胸腺嘧啶) Zidovudine Related Compound C (thymine) 对照品/标准品1724521 齐多夫定杂质B Zidovudine Related Compound B 对照品/标准品1724500 齐多夫定 Zidovudine 对照品/标准品1724317 扎西他滨杂质A Zalcitabine Related Compound A 对照品/标准品1724306 扎西他滨 Zalcitabine 对照品/标准品1724000 盐酸育亨宾 Yohimbine Hydrochloride 对照品/标准品1722005 木糖 Xylose 对照品/标准品1721002 盐酸赛洛唑啉 Xylometazoline Hydrochloride 对照品/标准品1720600 木糖醇 Xylitol 对照品/标准品1720429 盐酸赛拉嗪 Xylazine Hydrochloride 对照品/标准品1720407 赛拉嗪 Xylazine 对照品/标准品1720203 呫吨酮 Xanthone 对照品/标准品1720000 呫吨酸 Xanthanoic Acid 对照品/标准品1719102 华法林杂质 A Warfarin Related Compound A 对照品/标准品1719000 华法林 Warfarin 对照品/标准品1717708 牡荆素(牡荆甙) Vitexin 对照品/标准品1717504 含量测定系统适用性用维生素D Vitamin D Assay System Suitability 对照品/标准品1716002 维生素A Vitamin A 对照品/标准品1715000 硫酸紫霉素 Viomycin Sulfate 对照品/标准品1714528 长春瑞滨杂质A Vinorelbine Related Compound A 对照品/标准品1714506 酒石酸长春瑞滨 Vinorelbine Tartrate 对照品/标准品1714007 硫酸长春新碱 Vincristine Sulfate 对照品/标准品1713004 硫酸长春碱 Vinblastine Sulfate 对照品/标准品1711508 阿糖腺苷 Vidarabine 对照品/标准品1711472 维替泊芬杂质A Verteporfin Related Compound A 对照品/标准品1711461 维替泊芬 Verteporfin 对照品/标准品1711440 维拉帕米杂质F Verapamil Related Compound F 对照品/标准品1711439 维拉帕米杂质E Verapamil Related Compound E 对照品/标准品1711428 维拉帕米杂质D Verapamil Related Compound D 对照品/标准品1711406 维拉帕米杂质B Verapamil Related Compound B 对照品/标准品1711304 维拉帕米杂质A Verapamil Related Compound A 对照品/标准品1711224 维库溴铵杂质F Vecuronium Bromide Related Compound F 对照品/标准品1711202 盐酸维拉帕米 Verapamil Hydrochloride 对照品/标准品1711188 维库溴铵杂质C Vecuronium Bromide Related Compound C 对照品/标准品1711177 维库溴铵杂质B Vecuronium Bromide Related Compound B 对照品/标准品1711166 维库溴铵杂质A Vecuronium Bromide Related Compound A 对照品/标准品1711155 维库溴铵 Vecuronium Bromide 对照品/标准品1711133 赖氨加压素 Lypressin 对照品/标准品1711100 加压素 Vasopressin 对照品/标准品1711009 香草醛熔点标准品 Vanillin Melting Point Standard 对照品/标准品1710006 香草醛 Vanillin 对照品/标准品1709018 Vancomycin B with Monodechlorovancomycin 对照品/标准品1709007 盐酸万古霉素 Vancomycin Hydrochloride 对照品/标准品1708795 缬沙坦杂质 C Valsartan Related Compound C 对照品/标准品1708784 缬沙坦杂质 B Valsartan Related Compound B 对照品/标准品1708773 缬沙坦杂质 A Valsartan Related Compound A 对照品/标准品1708762 缬沙坦 Valsartan 对照品/标准品1708751 戊柔比星分离度用混合物 Valrubicin Resolution Mixture 对照品/标准品1708730 戊柔比星 Valrubicin 对照品/标准品1708729 丙戊酸杂质A Valproic Acid Related Compound A 对照品/标准品1708718 丙戊酸杂质 B Valproic Acid Related Compound B 对照品/标准品1708707 丙戊酸 Valproic Acid 对照品/标准品1708503 L- 缬氨酸 L-Valine 对照品/标准品1708015 D-缬更昔洛韦 D-Valganciclovir 对照品/标准品1708004 缬更昔洛韦盐酸盐 Valganciclovir Hydrochloride 对照品/标准品1707908 缬草烯酸 Valerenic Acid 对照品/标准品1707894 万乃洛韦杂质G Valacyclovir Related Compound G 对照品/标准品1707883 万乃洛韦杂质F Valacyclovir Related Compound F 对照品/标准品1707872 万乃洛韦杂质E Valacyclovir Related Compound E 对照品/标准品1707861 万乃洛韦杂质D Valacyclovir Related Compound D 对照品/标准品1707855 万乃洛韦杂质C Valacyclovir Related Compound C 对照品/标准品1707839 盐酸万乃洛韦 Valacyclovir Hydrochloride 对照品/标准品1707806 熊去氧胆酸 Ursodiol 对照品/标准品1706701 C13尿素 Urea C 13 对照品/标准品1706698 尿素 Urea 对照品/标准品1706009 乌拉莫司汀 Uracil Mustard 对照品/标准品1705800 阿糖尿苷 Uracil Arabinoside 对照品/标准品1705505 十一烯酸 Undecylenic Acid 对照品/标准品1705323 泛癸利酮杂质A Ubidecarenone Related Compound A 对照品/标准品1705312 系统适用性试验用泛癸利酮 Ubidecarenone for System Suitability 对照品/标准品1705301 泛癸利酮 Ubidecarenone 对照品/标准品1705006 L- 酪氨酸 L-Tyrosine 对照品/标准品1704003 泰洛沙泊 Tyloxapol 对照品/标准品1703850 酒石酸泰洛星 Tylosin Tartrate 对照品/标准品1703805 泰洛星 Tylosin 对照品/标准品1702008 氯筒箭毒碱 Tuboc
瘦肉精莱克多巴、盐酸克伦特罗检测瘦肉精莱克多巴、盐酸克伦特罗检测仪器高效液相色谱仪,紫外检测器超声波振动器溶剂过滤器离心机分析天平恒温摇床固相萃取装置,带萃取小柱匀浆机氮吹仪试剂乙腈:色谱纯磷酸二氢钾:分析纯磷酸甲醇:色谱纯乙醚;分析纯正己烷:分析纯盐酸溶液乙酸缓冲液NaOH溶液超纯水标准品:莱克多巴、盐酸克伦特罗样品制备 看到上面的仪器和试剂,大家可能也会看出前处理的方法了。不管的饲料还是肉制品样品的制备,过程一般都是称取,粉碎(匀浆),溶解,超声提取,离心提取,过滤,固相萃取,吹干,过滤等。只是在提取时样品完全提取可能比较困难,提取条件一定得控制好,比如温度、振动、时间(如果其它前处理设备性能不够理想的话,处理时间尽量长一点)、提取试剂等因数的控制和把握。色谱条件色谱柱:Venusil CN ,4.6mm×250mm,5µm流动相:乙腈:0.01mol/L磷酸二氢钾(用磷酸调节pH值至3.0)=30:70(V:V)检测波长:215nm流速:1.0mL/min柱温:室温进样量:20ul标准品色谱图:http://ng1.17img.cn/bbsfiles/images/2013/12/201312281531_485166_2369266_3.png 分析实验讲的就是准确、可靠,准确可靠的方法就是好方法。实验过程中我们一般没必要把实验做的最好,但我们可以通过优化色谱条件把实验尽量最好。

盐酸洛美利嗪含量测定方法研究本品为二苯哌嗪类钙通道阻滞剂,具有选择性的脑血管舒张作用。毒理研究遗传毒性:微生物回复突变试验、染色体畸变试验和小鼠微核试验结果均为阴性。下面主要针对盐酸洛美利嗪的含量测定方法进行研究。 一、容量法洛美利嗪为有机碱,可与高氯酸发生酸碱中和反应。1.指示剂选择和滴定终点的确定精密称取盐酸洛美利嗪约0.2g,加入15ml冰醋酸,振摇使溶解,加入5ml醋酸酐及5ml醋酸汞试液,加入1滴结晶紫指示液,并用电位计指示电位的变化,描绘滴定曲线。试验证明,当电位发生突跃时,溶液呈黄绿色。以高氯酸滴定液(0.1mol/L)滴定,并将滴定的结果用空白校正。每1ml高氯酸滴定液(0.1mol/L)相当于27.073mg的盐酸洛美利嗪。2.指示剂滴定法与电位滴定法含量测定的结果比较精密称取10份样品,每份约0.2g,加入15ml冰醋酸,振摇使溶解,加入5ml醋酸酐及5ml醋酸汞试液,加入1滴结晶紫指示液,其中五份做电位法滴定,另外五份做指示剂法确定终点,分别计算含量,数据见表1,从数据可知,电位法和指示剂法结果基本一致。用指示剂指示终点的三批样品的结果见表1。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251617_415393_2583865_3.jpg3.重复性试验及中间精密度试验三天内对同一批样品分别按80%、100%、120%三个水平各称取二份,指示剂滴定法测定其含量,结果见表2, 结果表明本法重复性及精密度较好。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251618_415394_2583865_3.jpg二、高效液相色谱法(HPLC)1.色谱条件及系统适用性试验(1)色谱条件:色谱柱:以十八烷基硅烷键合硅胶为填充剂(Xtimate C18),250×4.6mm,5um。流动相:甲醇-0.03mol/L磷酸氢二钾缓冲液(用磷酸调节pH4.0)(85:15),使用前经0.45μm有机滤膜抽滤并脱气。检测波长:225nm流速:1.0ml/min进样体积:20μl(2)系统适用性试验:精密称取干燥恒重的对照品约25mg置50ml量瓶中,用流动相溶解并稀释至刻度,摇匀作为贮备液。精密量取贮备液5.0ml置50ml量瓶中,精密量取20ml注入液相色谱仪,记录色谱图,连续进样6次,计算精密度。结果见表3。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251619_415395_2583865_3.jpg由试验结果可知,RSD小于1%,表明该色谱条件下精密度良好,系统适用性符合规定。2.线性关系精密称取干燥恒重的对照品约25mg置50ml量瓶中,用流动相溶解并稀释至刻度,摇匀作为贮备液。精密量取贮备液3.0、4.0、5.0、6.0、7.0和8.0ml置50ml量瓶中,用流动相稀释定容,摇匀作为溶液1、2、3、4、5和6,各精密量取20μl注入液相色谱仪。以标准溶液的浓度作为横坐标,色谱峰峰面积为纵坐标,绘制标准曲线。结果见表4。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251620_415396_2583865_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/12/201212251612_415391_2583865_3.jpg3.含量测定方法及测定结果精密称取本品适量,用流动相制成每1ml中约含50mg盐酸洛美利嗪的溶液,作为供试品溶液。另称取经恒重的对照品,同法制成每1ml中约含50mg对照品溶液。按前述高效液相色谱条件,分别量取对照品溶液和供试品溶液各20ml注入色谱仪,记录色谱图,按外标法计算含量。三批样品的HPLC法含量测定结果见表7-16。三批样品的含量测定结果见表5. http://ng1.17img.cn/bbsfiles/images/2012/12/201212251620_415397_2583865_3.jpg三、结果讨论分别采用容量法和高效液相色谱法测定三批样品的含量,可以看出两种方法的准确度、精密度等均能满足盐酸洛美利嗪含量检测的要求。其中容量法相对简单,系统误差小,故采用容量法作为含量测定的方法。
原料药盐酸米多君标准(国内:YBH09452006),5-氨基乙酰丙酸盐酸盐标准。若有特殊要求也可站内信息联系!有国外标准也行。
参考美国药典液相色谱条件,用YMC-Triart C8色谱柱测定盐酸奥洛他定滴眼液的含量,系统适应性试验中理论塔板数、拖尾因子、相对标准偏差等均符合规定。
在重金属元素分析中,我们不可能忽略的一个条件就是标准品的酸度问题,通常情况下,金属元素的标准品都是溶解在酸性介质中,比如5%的硝酸溶液中。无论在[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]还是在原子荧光光谱分析过程中,都要非常重视酸度。有一次我做铅的检测,在标准序列的配置过程中,我贪图省事,没有逐级用酸进行稀释,导致做标准曲线的时候,铅已经发生水解,形成了沉淀。铅水解的化学方程式如下:[align=center]Pb[sup]2+[/sup]+2H[sub]2[/sub]O=Pb(OH)[sub]2[/sub]+2H[sup]+[/sup][/align]只有在酸度条件合适的情况下,才能抑制铅的水解。很明显,我这次的检测结果是失败的,连标准品的线性都没有。忽略了酸度的影响,其结果只能是毫无线性可言。我查阅了相关资料,发现除轻金属外,一般情况下,各种重金属都要在酸溶液中进行妥善保存。从这一点上,我看到了如下几点:一、 标准物质控制的严谨性。在标准溶液的生产和使用过程中,都要严格按照标准和规程进行稀释。标准物质绝对不是简简单单溶解到水里面了事,就是标准物质的盐溶液也存在着水解效应。标准物质要考虑的事情不单单是配置成为溶液,溶液要保持稳定性,只有稳定的溶液才能作为检测的基准。二、 标准物质质量的统一性。标准物质在生产的过程中要考虑到溯源性,必须量值进行合理的传递。从原料关,溶液关都要严格把关。最终的结果是量值溯源要溯源到国家计量院。标准物质是一种严肃的物质。三、 标准物质生产的严肃性。标准物质不是简简单单的盐溶液,整个生产过程中要格外严谨,这就要求生产者有丰富的经验和严谨求实的作风。标准物质生产过程还必须进行合理的质检。同一批次要验证均匀性,不同批次要验证重复性。标准品证书会给出生产日期、不确定度、保存条件等,可以标准品证书同样很重要。标准物质的出现极大的方便了我们的检测,但是,在标准品使用过程中,我认为应该注意以下几个问题:一、 严格按照保存条件进行保存,尤其是农药残留类的标准品,因为农残的种类多,往往我们会顾此失彼,这是标准品控制过程中的难点。一旦标准品失效或者激昂接我们的检测结果就会失实。二、 定容过程要严谨。标准品开封以后要妥善保存。同时,也要仔细分析国家标准中,为什么要这么配置标准溶液。我认为通过深入领会标准,也可以防风险。三、 好好看标准品证书。仔细分析标准物质的物理性质和化学性质,这样才能提升我们的水平,进而降低检测过程中的风险。标准品方便了我们的检测的过程,但是也还是要注意学会如何使用标准品,相比于按照标准进行配置,使用标准物质绝对不是万事大吉。否则,存在的检测风险依然很大。[sup] [/sup]
这个问题涉及到很多方面,故放在综合区,望版主谅解。近日在考察注射用盐酸阿糖胞苷是否可用USP37的标准。发现一个问题,USP37收载的该剂型的活性物质是阿糖胞苷,而不是盐酸阿糖胞苷;而在查阅国外的几个制药公司的该产品的说明书,注明的活性物质也是阿糖胞苷原型,而非盐酸盐。故在此想请教对药物化学和药物分析很了解的童鞋们几个问题:1、国外的该产品说明书上注明的是阿糖胞苷原型药,USP37收载的也是原型药,是否说明在国外该制剂采用的原料药是阿糖胞苷,而非其盐酸盐?2、国内的制药厂家的该产品说明书上注明的活性物质是盐酸阿糖胞苷,且中国药典收载的也是盐酸阿糖胞苷,毫无疑问说明原料药采用的是盐酸阿糖胞苷,为什么要用盐酸盐,而不跟国外一样采用原型药?是为了避开专利还是盐酸盐的形式更有利于人体吸收?3、USP37收载原型药,被检测的药物是盐酸盐,是否说明该药物不能用USP37的标准来检?4、在哪里可以查到国外药物的专利,及其详细信息,如药物的化学结构、化学式、其专利到期的时间等?5、如何可以准确地知道国外一些药品制剂所使用的原料的详细信息?尤其是理化方面的信息,这些信息在说明书上体现的很少。望高手不吝赐教!谢谢!
请教,,在高温(温度90c)盐酸溶液体系下(盐酸10%上下)的矿浆浸液中。怎样测量它的溶液-酸度-和-氯离子浓度-等。。。有什么快速准确的仪器或方法来测量高酸,高氯离子浓度溶液的设备。。。。。。







 我要推广仪器
我要推广仪器
 下载APP
下载APP