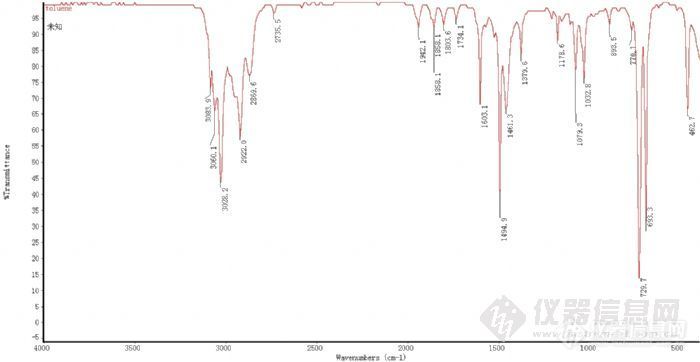
今天拿到一张苯标准品、一张甲苯标准品的IR谱图。想请问一下,里面的吸收峰都是什么振动引起的呢?峰该怎样归属呢?谢谢大家~这张是苯的标准图:http://ng1.17img.cn/bbsfiles/images/2011/06/201106211618_300779_1905813_3.jpg这张是甲苯的标准图:http://ng1.17img.cn/bbsfiles/images/2011/06/201106211618_300780_1905813_3.jpg
有人有间溴甲苯醚含纯的色谱分析方法或者间溴甲苯醚的相关标准吗?急切需要帮助,谢谢各位大侠了。
做胶粘剂中苯、甲苯、二甲苯,卤代烃等的含量,所用苯、甲苯、二甲苯,卤代烃是否为标准品?还是色谱纯的试剂即可??谢谢!!!
求助:国外食品包装或食品中关于甲苯的标准?哪位大侠知道,请不吝赐教,在下万分感谢
连、偏、均三甲苯的标准品,要有标准品证书的在哪里可以购买?
大家好,我们买了活性炭管中苯甲苯二甲苯成份分析标准物质GBW(E)080237、GBW(E)080238,证书不知道怎么弄得,丢了,呜呜,是中国疾控中心职业卫生与中毒控制所生产地,请问大家买过吗?标准值是多少啊?
我现在依据国家职业卫生标准GBZ/T160.42-2004做苯、甲苯、苯胺的工作场所空气中有毒物质的分析,做不好标准曲线,请问哪位高手有电子版曲线图上传一份,谢!谢!
[font=宋体][size=14px][/size][/font][font='微软雅黑','sans-serif']盐酸中甲苯测定,可以直接萃取后进样分析吗?标准样品配制需要在盐酸中加入不同量的甲苯配制吗?[/font][font='微软雅黑','sans-serif']求助问题来自微信群。[/font][font=宋体][size=14px][/size][/font]
最近刚接触涂料(各类漆)中有害物质分析,主要是测定其中含有的苯、甲苯、二甲苯含量,依照GB18581-2001 中气谱(内标法)分析,但遇到些问题,希望各位高手帮忙指点一下~~先按照GB配置含有苯、甲苯、二甲苯的标准溶液,得到谱图,得到相关的三苯保留时间,同时计算出校正因子,接下来进行样品分析,问题出现了,如何准确判断出样品中是否含有三苯呢,是依靠与内标物的相对保留时间吗?那样品保留时间与标样保留时间相差多少才能确定样品中是否含有三苯呢?因为现在漆中成分复杂,所以很头痛,希望大家指点一下~~
购得的有机氯标准品是以甲苯为溶剂,想要配成有机氯的工作标准溶液(水溶液)进行固相萃取,请教如何配制?先将标准品溶于甲醇后,再配成水溶液可行吗?
[color=#444444]最近在做残留分析实验,咨询一下大师们关于农药苯噻酰草胺,苄嘧磺隆,吡嘧磺隆在高效液相色谱分析时最佳的色谱柱及条件?在这里先表示真挚的感谢![/color]
求混合二甲苯的分析方法
关于脂肪酸标准品甲苯液的保存一直困惑着我,想看哪位老师能指点一二。 我是检婴幼儿乳粉的,天天都得检脂肪酸含量。我用的是国标一法(甲醇甲酯化法)。首先,我不知道是不是应该每次检的时候都进一下标准品?其次,不知道为什么每天标准品出的峰积不太一样,有时在误差允许范围,有时候我觉得偏的离谱,都把我搞糊涂啦,不知道是依哪个为标准啦?再者,处理好的标准品不知道该怎么保存?不知道过多久就失效啦? 请知道的老师教教我,我也只能简单的说声谢谢!
配置PBB,PBDE,以及PAHS的标准溶液能否用甲苯的分析纯来稀释?
请教:FID分析甲苯、二甲苯,灵敏度怎么这么差呀。我买的国家标准物质,用甲醇稀释至20ug/ml,进样量1ul,分流比10:1,结果峰面积只有1000多uv.sec,很小。不分流的话溶剂峰太大了把目标峰就盖了。
我们想做二甲苯的标准曲线,实验室只有一瓶二甲苯的色谱纯,拿它当标准品用,包装上面没写浓度,只写了含量99.8%,请问这样怎么算出它的体积浓度或者质量浓度呢?谢谢!
我想分析胶粘剂中残留甲苯的量,可以用顶空法测试吗?标准液应如何配制?
买的苯系物混合标准液里面没有甲苯,怎么回事?
我想分析胶粘剂中残留甲苯的量,可以用顶空法测试吗?标准液应如何配制
我们想做二甲苯的标准曲线,实验室只有一瓶二甲苯的色谱纯,拿它当标准品用,包装上面没写浓度,只写了含量99.8%,请问这样怎么算出它的体积浓度或者质量浓度呢?谢谢!
请教各位大侠,俺现在分析汽油中的苯、甲苯(sh/t 0713-2002),因汽油中含有甲醇(约1%),采用标准所述填充柱系统,甲醇干扰苯的出峰;改用毛细柱系统不干扰。这是在做标样时的情况。 但是分析汽油样品时实际样品中的低沸点组分还是干扰苯的岀峰,分离效果不好。请教请教![em09508]
二硫化碳解析苯系物,万万没想到分析纯的二硫化碳里面含有苯和甲苯,标准曲线怎么做呀?我该怎么做标曲,我用的是外标法
甲苯中甲基汞的标准品用GB5009.14法检测,标准品用什么溶剂溶解?望老师不吝赐教
用GC分析空气中气态甲苯的浓度,怎么做标准曲线?我们没有甲苯标气,用分析纯的液态甲苯配置标液可以么?如果可以的话,选用什么溶剂来稀释液态甲苯比较合适?这样做对结果有影响么? 补充,我们用的是填充柱,FID检测器,待测气态甲苯浓度在500ppm左右。 希望各位高手多多指教,小女子在这里先谢过啦!非常着急啊!
一待分析混合溶液,对二甲苯含量在99%左右,其中的杂质有甲苯,邻位二甲苯,间位二甲苯等,具有这些杂质的纯溶液,如果分析这些杂质的含量,能否用待分析混合溶液作为标准物质的溶剂做标准曲线呢?还是应该选择一个不含待分析杂质的有机试剂作为溶剂?为什么有的人说可以用待分析的混合溶液可以作为溶剂做标准曲线呢,我觉得不对呢,因为里面含有待测的杂质啊,是不可以的吧?如果用其它溶剂,用哪一样比较合适呢?正己烷?正壬烷?还是其它?如果用其它的溶剂,会不会存在基体影响?造成基体差别?多谢大家哦~~~~
6.2.2用标准溶液绘制标准曲线于3个50mL容量瓶中,先加入少量二硫化碳,用10μL注射器准确量取一定量的苯、甲苯和二甲苯分别注入容量瓶中,加、硫化碳至刻度,配成一定浓度的贮备液。临用前取一定量的贮备液用二硫化碳逐级稀释成苯、甲苯和二甲苯含量为0.005,0.01,0.05,0.2μg/mL的混合标准液。分别取1μL进样,测量保留时间及峰高,每个浓度重复3次,取峰高的平均值,以苯、甲苯和二甲苯的含量(μg/μL)为横坐标,平均峰高(mm)为纵坐标,绘制标准曲线。并计算回归线的斜率,以斜率的倒数Bg[μg/(mL• mm)]作样品测定的计算因子。6.2.3 测定校正因子当仪器的稳定性能差,可用单点校正法求校正因子。在样品测定的同时,分别取零浓度和与样品热解吸气(或二硫化碳提取液)中含苯、甲苯和二甲苯浓度相接近时标准气体1mL或标准溶液1μL按6.2.1或6.2.2操作,测量零浓度和标准的色谱峰高(mm)和保留时间,用式(1)计算校正因子。¦ =cs/(hs-h0)………………………………………(1)式中: ¦ 一一校正因子,μg/(mL• mm)(对热解吸气样)或μg/(μL• mm)(对二硫化碳提取液样);cs一一标准气体或标准溶液浓度,μg/mL或μg/μL;h0、hs-一零浓度、标准的平均峰高,mm。6.3 样品分析6.3.1热解吸法进样将已采样的活性炭管与100mL注射器相连,置于热解吸装置上,用氮气以50~60mL/min的速度于350℃下解吸,解吸体积为100mL,取1mL解吸气进色谱柱,用保留时间定性,峰高(mm)定量。每个样品作三次分析,求峰高的平均值。同时,取一个未采样的活性炭管,按样品管同样操作,测定空白管的平均峰高。6.3.2 二硫化碳提取法进样将活性炭倒入具塞刻度试管中,加1.0mL二硫化碳,塞紧管塞,放置1h,并不时振摇,取1μL进色谱柱,用保留时间定性,峰高(mm)定量。每个样品作三次分析,求峰高的平均值。同时,取一个未经采样的活性炭管按样品管同样操作,测量空白管的平均峰高(mm)。7 结果计算7.1 将采样体积按式(2)换算成标准状态下的采样体积。V0=Vt• T0/(273+t)• p/p0………………………………………(2)式中:V0――换算成标准状态下的采样体积,L;Vt――采样体积,L;T0――标准状态的绝对温度, 273K;t――采样时采样点的温度,℃;p0――标准状态的大气压力, 101.3kPa;p――采样时采样点的大气压力, kPa。7.2 用热解吸法时,空气中苯、甲苯和二甲苯浓度按式(3)计算。C=(h-h0)Bg/(V0• Eg)×100…………………………………(3)式中:c――空气中苯或甲苯、二甲苯的浓度,mg/m3;h――样品峰高的平均值, mm;h0――空白管的峰高, mm;Bg――由6.2.1得到的计算因子,μg/(mL• mm);Eg――由实验确定的热解吸效率。7.3 用二硫化碳提取法时,空气中苯、甲苯和二甲苯浓度按式(4)计算。C=(h-h0)• Bs/(V0• Es)×1000………………………………(4)式中:c一一苯或甲苯、二甲苯的浓度,mg/m3;Bs一一由6.2.2得到的校正因子,μg/(μL• mm);Es一一由实验确定的二硫化碳提取的效率。7.4 用校正因子时空气中苯、甲苯、二甲苯浓度按式(5)计算。C=(h-h0)• ¦ /(V0• Eg)×100或C=(h-h0)• ¦ /(V0• Es)×1000…………(5)式中:¦ ――由6.2.3得到的校正因子,mg/(mL• mm)(对热解吸气样)或μg/(μL• mm)(对用二硫化碳提取液样)。8 精密度和准确度8.1精密度8.1.1用热解吸法苯浓度为0.1,0.5和2.0μg/mL的气样,重复测定的变异系数分别为7%,6%和4%,甲苯浓度为0.1,0.5和2.0μg/mL气样,重复测定的变异系数为9%,7%和4%,二甲苯的浓度为0.1,0.5和2.0μ/mL气样,重复测定的变异系数为9%,6%和5%。8.1.2用二硫化碳提取法苯的浓度为8.78和21.9μg/mL的液体样品,重复测定的变异系数为7%和5%,甲苯浓度为17.3和43.3μg/mL液体样品,重复测定的变并系数分别为5%和4%,二甲苯浓度为35.2和87.9μg/mL液体样品,重复测定的变异系数为5%和7%。8.2准确度用热解吸法对苯含量为5,50和500μg的回收率分别为96%,97%和97%,甲苯含量为10,100和1000μg的回收率分别为90%,91%和94%,二甲苯含量95.511g的回收率为82%;二硫化碳提取法,对苯含量为0.5,21.1和200μg的回收率分别为95%,94%和91%,甲苯含量为0.5,41.6和500μg的回收率分别为99%,99%和93%,二甲苯含量为0.5,34.4和500μg的回收率分别为101%,100%和90%。 附录A二硫化碳的纯化方法(补充件)二硫化碳用5%的浓硫酸甲醛溶液反复提取,直至硫酸无色为止,用蒸馏水洗二硫化碳至中性再用无水硫酸钠干燥,重蒸馏,贮于冰箱中备用。附录B[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析空气中苯、甲苯和二甲苯的实例(参考件)B1 色谱条件B1.1 色谱柱温度:90℃;B1.2 检测室温度:150℃;B1.3 汽化室温度:150℃;B1.4 载气:氮,50mL/min。B2 用B1的色谱条件操作的色谱图见图B1和图B2。附加说明:本标准由卫生部卫生监督司提出。本标准由广东省职业病防治院、杭州市卫生防疫站、南京市卫生防疫站、沈阳市卫生防疫站负责起草。本标准主要起草人叶能权、陆展荣、童映芳。本标准由卫生部委托技术归口单位中国预防医学科学院环境卫生监测所负责解释。
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定碳九加氢产品中三甲苯含量的测定方伟民宁波广昌达新材料有限公司摘要:建立了用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]、DB-FFAP色谱柱分析碳九加氢产品中各三甲苯含量的方法。该方法采用单色谱柱、单氢火焰离子化检测器(FID),分流进样分析,采用校正面积归一化法对分析结果进行定量。该方法的特点是仪器配置简单,样品用量少,分离效果显著,结果准确。关键词:[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]DB-FFAP色谱柱碳九加氢三甲苯前言随着国内乙烯工程的大量投入使用,裂解制乙烯的副产品乙烯胶油大量产出,这些副产品因含有大量的九个碳原子的芳烃馏分,统称碳九芳烃。主要组分有异丙苯、正丙苯、乙基甲苯、均三甲苯、偏三甲苯、邻三甲苯、茚等。碳九芳烃馏分组分复杂,沸点相近,难以一一分离,目前主要分离出偏三甲苯和均三甲苯用于制偏苯三酸酐和均苯四甲酸二酐等,用于涂料,合成树脂等。对这些副产品进行加氢再利用,加氢后的产品中三甲苯的含量的高低直接影响产品销售对象。因此建立分析加氢产品中三甲苯含量的方法尤其重要。本方法仪器配置简单,样品用量少,分离效果显著,结果准确,可用于工厂常规分析。1 实验部分1.1 试剂与仪器设备1.1.1 试剂试剂名称 规格 生产厂家高纯氮气 纯度≥99.999% 宁波亚大气体有限公司高纯氢气 纯度≥99.999% 宁波亚大气体有限公司压缩空气 —— 现场装置直供邻二甲苯 纯度:99.9% 阿拉丁试剂1,3,5-三甲苯 纯度:89.0% 阿拉丁试剂1,2,4-三甲苯 纯度:99.5% 阿拉丁试剂1,2,3-三甲苯 纯度:99.9% 阿拉丁试剂1,2,4,5-四甲苯 纯度:90.0% 阿拉丁试剂1,2,3,5-四甲苯 纯度:98.0% 阿拉丁试剂1.1.2仪器和设备1.1.2.1 电子天平:赛多利斯BSA124S电子天平,精确到0.0001g1.1.2.2 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:常州盘诺仪器有限公司的A90[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],配置有氢火焰离子化检测器(FID),B75型16位自动进样器,10μL进样针,1.1.2.3 色谱柱:DB-FFAP,60×0.25×0.50,J&W1.2 色谱操作条件选择及探讨1.2.1载气的选择由于反应体系中异构体较多,选择载气主要从柱效考虑用氢火焰离子化检测器一般多用氦气和氮气做载气。用氦气做载气,有点可以缩短分析时间,但分析成本较高,选用氮气做载气,柱效高,分离效果好,但分析时间较长,综合考虑选用氮气做载气。1.2.2色谱柱的选择三甲苯与四甲苯的同分异构体比较多,各同分异构体的沸点如表1:表1三甲苯和四甲苯各同分异构体的沸点组分 1,3,5-三甲苯 1,2,4-三甲苯 1,2,3-三甲苯 1,2,4,5-四甲苯 1,2,3,5-四甲苯 1,2,3,4-四甲苯沸点/℃ 164.7 169.4 176.1 196.8 199.0 205.0从表1中我们可以看出三甲苯和四甲苯的同分异构体的沸点非常接近,特别是三甲苯的三种同分异构体。要使组分完全分离需选用强级性的分离柱,如FFAP毛细管柱、OV101毛细管柱、SE-30毛细管柱、聚乙二醇-20等色谱柱。1.2.3色谱柱及色谱条件从分析要求、分析效果和分析方法的经济消耗等方面进行考虑选用如下的色谱柱及操作条件。如表2:表2色谱柱及色谱条件色谱柱固定相 聚乙二醇TPA,极性固定相柱长/m 60柱内径/mm 0.25固定液膜厚度/μm 0.50载气 氮气检测器 检测器类型 氢火焰离子化检测器 温度/℃ 250 空气流量/(mL/min) 400 氢气流量/(mL/min) 30汽化室 温度/℃ 230 分流比 100:1柱箱 初温/℃ 80 初温保持时间/min 2 一阶升温速率/(℃/min) 4 一阶终止温度/℃ 136 一阶温度保持时间/min 0 二阶升温速率/(℃/min) 2 二阶终止温度/℃ 148 二阶温度保持时间/min 0 三阶升温速率/(℃/min) 2 三阶终止温度/℃ 166 三阶温度保持时间/min 0 四阶升温速率/(℃/min) 5 四阶终止温度/℃ 230 四阶温度保持时间/min 3柱流量/(mL/min) 1.5进样量/μL 0.42 结果与讨论2.1 色谱图中组分的定性在相同实验条件下对标准物质对各组分进行分析,采用保留时间定性,用标准物质配制混合标样进行分析结果如图1。各组分都能在色谱图上出峰,且分离效果明显。 图1 混合标准物质在色谱上的色谱图1)1,3,5-三甲苯 2) 1,2,4-三甲苯 3) 1,2,3-三甲苯4) 1,2,4,5-四甲苯 5)1,2,3,5-四甲苯2.2 定量分析样品中的各物质都能在色谱图上出峰,所以采用校正面积归一化法进行定量。用称量法配制含1,3,5-三甲苯、1,2,4-三甲苯、1,2,3-三甲苯、1,2,4,5-四甲苯、1,2,3,5-四甲苯和邻二甲苯,用邻二甲苯做标准物质计算其它组分的相对质量校正因子。各组分称量应精确至0.0001g,含量计算应精确至0.0001%(质量百分数)。并按下式进行计算: fi—标样中i相对邻二甲苯的质量校正因子A— 标样中邻二甲苯的峰面积Ai—标样中组分i的峰面积m—标样中邻二甲苯的质量,gmi—标样中组分i的质量,g2.2.1 校正因子的测定用邻二甲苯做标准物质测定各组分的质量相对校正因子,其结果如下:表3 各组分重复质量校正因子的测定标样 组分 称重(g) 含量(%) 次数 峰面积 校正因子 1,3,5-三甲苯 2.2643 9.0897 1 7114.08 0.99 2 6190.29 0.99 3 6313.75 1.00 1,2,4-三甲苯 9.0575 36.5057 1 28312.47 1.00 2 2463.46 1.01 3 2511.18 1.00 1,2,3-三甲苯 1.1245 4.5094 1 3676.47 0.95 2 3199.86 0.96 3 3263.33 0.96 1,2,4,5-四甲苯 1.2738 5.1083 1 4123.67 0.96 2 3578.28 0.97 3 3681.98 0.96 1,2,3,5-四甲苯 0.2218 0.8894 1 675.38 1.02 2 586.63 1.03 3 603.26 1.02 邻二甲苯 10.7673 43.1785 1 33396.17 1.00 2 29238.58 1.00 3 29511.96 1.00表4 校正因子测定结果分析组分 实验1 实验2 实验3 平均值 相对偏差(%)1,3,5-三甲苯 0.99 0.99 1.00 0.99 1.011,2,4=三甲苯 1.00 1.01 1.00 1.00 1.001,2,3-三甲苯 0.95 0.96 0.96 0.96 1.041,2,3,5-四甲苯 0.96 0.97 0.96 0.96 1.041,2,4,5-四甲苯 1.02 1.03 1.02 1.02 0.98在相同的测试条件下,连续三次测量标准混合液,计算各组分的质量校正因子的偏差小于5%,符合分要求。2.2.2检测限为了确定各组分的检测限,通过对混合标样的分析,以色谱峰高度基线噪声的3倍做为标准计算检出限。用邻二甲苯做基液,配制样品浓度0.0005%左右,连续分析样品6次,取平均用于计算各组分的检测限,其结果如下:表5 各组分检测限计算组分 平均峰高 配制浓度(% m/m) 信号噪声 检测限(% m/m)1,3,5-三甲苯 0.32 0.0005 0.05 0.000231,2,4-三甲苯 0.24 0.0004 0.05 0.000251,2,3-三甲苯 0.30 0.0005 0.05 0.000251,2,4,5-四甲苯 0.29 0.0005 0.05 0.000261,2,3,5-四甲苯 0.19 0.0003 0.05 0.000242.2.3方法验证和精密度分析用已知纯度的标准样品配制2组已知浓度的标准试样,在规定的实验方法条件下,用标样进行6次测试分析,6次的分析结果的平均值与该组分的理论值进行比对,确定方法的回收率和精密度,实验结果如表6:表6 标样回收和精密度计算结果组分 标样1# 配制浓度%(m/m) 平均值%(m/m) RSD,% 回收率,%1,3,5-三甲苯 4.1829 4.1746 2.82% 99.81,2,4=三甲苯 30.8511 30.5734 1.35% 99.11,2,3-三甲苯 2.3698 2.3390 2.52% 98.71,2,3,5-四甲苯 3.0255 2.9317 1.89% 96.91,2,4,5-四甲苯 1.1277 1.1085 1.05% 98.3组分 标样2# 配制浓度%(m/m) 平均值%(m/m) RSD,% 回收率,%1,3,5-三甲苯 6.5346 6.4889 2.68% 99.31,2,4=三甲苯 20.1885 20.0674 1.16% 99.41,2,3-三甲苯 4.3328 4.2158 2.15 % 97.31,2,3,5-四甲苯 8.1271 7.8589 2.57% 96.71,2,4,5-四甲苯 3.0554 3.0310 2.16% 99.2通过对标准混合样的分析,结果表明该方法相对偏差小于5%,满足分析要求。2.3实际样品分析在规定的色谱条件下,取罐样产品分析,其分析色谱图如下:图 2 G5209产品罐样色谱分析图表5 G5209产品罐样测定结果组分 结果%(m/m)1,3,5-三甲苯 5.71,2,4=三甲苯 32.51,2,3-三甲苯 3.21,2,3,5-四甲苯 2.91,2,4,5-四甲苯 0.82.4结论采用DB-FFAP色谱柱,进行分流进样,用校正面积归一化法分析碳九加氢产品中的三甲苯和四甲苯各同分异构体的含量,操作简单,分离效果好,分析结果稳定,准确度高。为产品进一步处理和销售提供了准确的可靠的分析数据。参考文献陶克毅.石油学报,1989,5(1):33~38SH/T 1773-2012 1,2,4-三甲苯纯度及烃类杂质的测定[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法傅若农,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]概论第二版
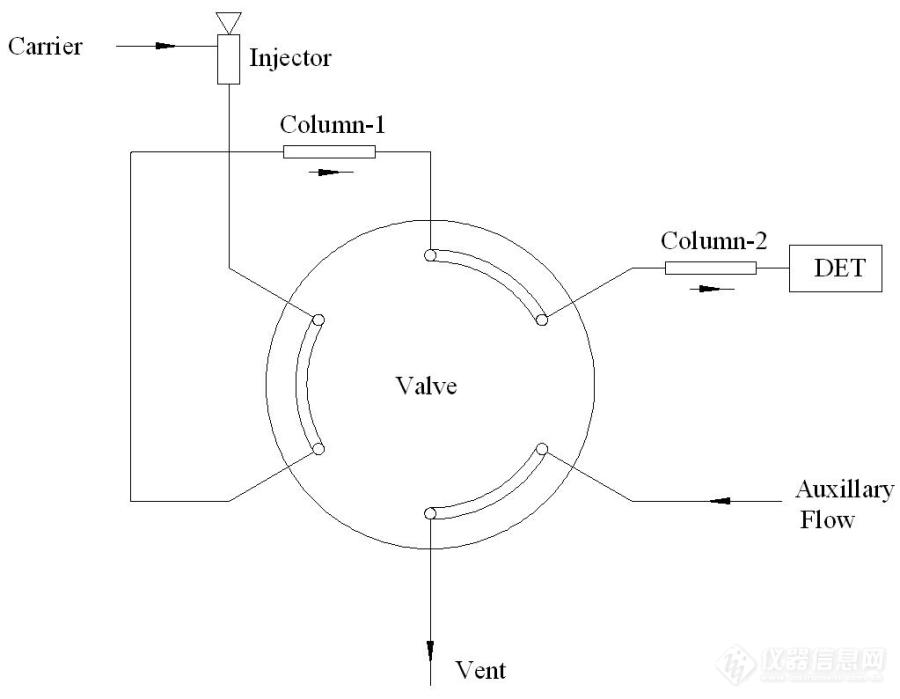
[color=black]SH/T0713 汽油中苯和甲苯分析系统的原理图解[/color][align=center][color=black]概述[/color][/align][color=black]《SH/T -0713-2002 车用汽油和航空汽油中苯和甲苯的含量测定法》基本原理解析。[/color][align=center][color=black]一 背景介绍[/color][/align][color=black]普通汽油中含有大量的(体积比30%-50%左右)的芳烃可提高汽油辛烷值,但其存在会增加汽车尾气中氮氧化物、一氧化碳、芳烃类等物质的排放量,其中苯和甲苯是有毒、有害物质,人体吸入后会使血液中白血球减少,免疫机能下降;亦是致癌物质,世界卫生组织和美国EPA认为人在接触1ug/m3的苯情况下,可使每百万人有4~8人患白血病的危险。[/color][color=black]随着环保意识的增强和汽油质量要求的升级,世界各国对汽油中苯含量 的要求均较严格,主要是因为苯为致癌物质,如果燃烧不完全会残存在汽车 尾气中,将会危害公众健康。[/color][color=black]石化行业标准《SH/T -0713-2002 车用汽油和航空汽油中苯和甲苯的含量测定法》目前为测定汽油中苯和甲苯含量的仲裁方法,在石油化工生产和检测行业中广泛应用。[/color][align=center][color=black]二 结构原理[/color][/align][color=black]SH/T 0713[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析系统结构如图1所示,系统由预切色谱柱(Column-1,标准方法要求使用非极性固定相的色谱柱)、自动六通阀、主分析柱(Column-2,标准方法要求使用强极性固定相的色谱柱)组成。通过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]系统分析程序对六通阀的精确定时切换,改变两根色谱柱的反吹和连接状态,实现汽油样品中苯和甲苯的分析。[/color][color=black]系统采用内标法定量,丁酮作为内标物准确添加于所有的内标标准品和所有待测样品中。[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109191858144515_4764_1604036_3.jpg[/img][/align][align=center][color=black]图1 SH/T 0713 硬件结构(系统待机状态)[/color][/align][color=black]系统的工作过程如下:[/color][color=black]待机和进样状态:[/color][color=black]汽油样品直接进样至[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的进样口(Injector)中,样品气化并进入预切色谱柱(Column-1),系统的简化结构如图2所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109191858147953_5747_1604036_3.jpg[/img][/align][align=center][color=black]图2 进样状态下系统结构简化示意图[/color][/align][color=black]预切色谱柱(Column-1)流出组分可能的谱图如图3所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109191858148734_5888_1604036_3.jpg[/img][/align][align=center][color=black]图3 预切柱流出组分色谱图[/color][/align][color=black]在预切色谱柱内,汽油中各组分大致按照分子量和沸点由小到大的顺序依次流出,苯、甲苯以及内标物(丁酮)保留较弱,与C6-C8多种烃类混合物一同出峰。三种物质流出顺序为丁酮、苯、甲苯。其中甲苯的保留时间最长,与C7-C8烃类混合物保留较为接近。[/color][color=black]切换反吹状态:[/color][color=black]当C8烃类物质流出预切色谱柱,C9烃类物质尚未流出时,自动六通阀转子旋转60度,系统状态变化为图4所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109191858149067_5565_1604036_3.jpg[/img][/align][align=center][color=black]图4 系统切换反吹状态[/color][/align][color=black]此时,本系统的简化结构如图5所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109191858151391_4198_1604036_3.jpg[/img][/align][align=center][color=black]图5 反吹状态下系统的简化结构示意图[/color][/align][color=black]预切色谱柱(Column-1)此时流量方向发生反向,色谱柱内的C9以上的重烃类物质被反吹流出色谱柱,经由Vent端口放空;C8以及C8以下的轻烃类物质(其中包含苯、甲苯、内标物——丁酮)进入主分析柱。在主分析柱的强极性固定相作用下,苯、甲苯和丁酮和轻烃类物质被分离开,系统谱图如图6所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109191858152172_4339_1604036_3.jpg[/img][/align][align=center][color=black]图6 系统谱图[/color][/align][color=black]系统复位[/color][color=black]当所有苯、甲苯和丁酮所有组分流出色谱柱后,六通阀再次旋转恢复至原始位置,如图1所示,本次分析完成。[/color][align=center][color=black]三 常见故障[/color][/align][color=black]切换点问题[/color][color=black]样品在预切柱中基本按照沸点排序,切换的时间需要选择在C8-C9烃类之间。切换点时间选择过短,会造成甲苯或者苯色谱峰面积的损失,切换点时间选择过长,会造成色谱图中干扰峰较多,对苯和甲苯的积分带来干扰。[/color][color=black]甲醇和乙醇的干扰[/color][color=black]《SH/T -0713-2002 车用汽油和航空汽油中苯和甲苯的含量测定法》分析标准中给出了三种配置色谱柱的方案,其中采用FFAP毛细管柱的方案,更加适合含甲醇或乙醇的车用汽油——甲醇或乙醇对分析结果干扰较小。[/color][color=black]定量[/color][color=black]本系统采用内标法定量,计算时内标量与样品量的确定比较重要。需要定期对标准曲线进行校准。[/color][align=center][color=black]小结[/color][/align][color=black]该分析系统长期运行后,需要对阀程序和定量操作进行定期校准。[/color]
我想要甲苯、乙苯、二甲苯(邻、间、对)的色谱标准样品(纯物质的那种,不带溶剂的),请问各位知不知道哪里有销售呢?
请问:测定废气中苯 甲苯 二甲苯的标准怎么配制?







 我要推广仪器
我要推广仪器
 下载APP
下载APP