还有个问题,hp-5的毛细柱做有机磷,氧化乐果为什么不出峰,试过很多次,标准品也是新打开的,不知为何,求解??? 谢谢
因为今天做甲胺磷 氧化乐果的时候 发现响应值比上次缩小了4倍,所以考虑衬管脏了,换了新衬管后,这两个峰却蒸发了,毒死蜱和甲基对硫磷有峰出,而且响应有所增高。
[table=100%][tr][td]本人用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检测农药氧化乐果标准物,色谱柱是用毛细管柱DB-1701P,操作条件为,柱温80℃,升温速率20℃/min,在260℃下保持3min,5℃/min,在270℃下保持1min。气化温度为220℃。测室温度为125℃。载气流速:恒流1.5mL/min(Psi)。氢气流速:80mL/min。空气流速:100mL/min。进样量为0.5ug。操作条件下,其他耗材和设备如衬管,堑圈均无污染,气源纯度均达到99.999%,单标和混标均不出峰,请问有什么因素会造成氧化乐果标准物不出峰?氧化乐果可能会在沸点135℃分解,这会不会在气化时已经分解导致不出峰且单标时色谱图是一直线。谢谢你们的专业意见![/td][/tr][/table]
最近气相做氧化乐果一直不出峰,不知道是标准品的问题还是仪器条件的原因,在网上查到氧化乐果不稳定,热易分解。不知道是不是这个原因分析原因有可能有以下几点,不知道大家觉得那种可能性比较大,或者大家有新的看法可以说说您的看法1、标准品分解的问题(这个原因可以排除,先后更换过好多次标液,新买的标液同样不出峰)2、柱子或者进样口衬管脏了,严重吸附造成。(色谱柱进样端截去一段,衬管更换新的,无济于事)3、进样口温度太高造成氧化乐果分解,不出峰(疑问,以前也是同样的温度出过峰)还有其他的原因吗?
请教:6890的FPD柱子衬管用一段时间之后,乙酰甲胺磷,氧化乐果标样出峰就不行,有时就没峰?什么原因?
用拉曼光谱检测了我的样品氧化层,我想知道里面到底有没有氧化铝,但是又没有氧化铝的拉曼标准峰位置让我对比。希望大家可以帮帮我。很着急。
求助环境空气五氧化二磷测定的钼蓝分光光度法,HJ546-2015的标准曲线,扩项的方法证实,没查到标准曲线的斜率要求,望做过的前辈提供一下对应浓度的吸光值对比一下。急急急,谢谢各位。
请问大家知道100ppm的甲胺磷、乙酰甲胺磷、氧化乐果的保存期限是多久吗?最近做农残标准曲线,使用的是去年8月份配的100ppm的标样,这三种农残0.1ppm的浓度基本没峰,1ppm的峰面积也明显较去年的小很多,不知是不是分解了。
氧化乐果低浓度不出峰,我用的是氮磷检测器的,高浓度能出峰 低浓度就不会出峰,请问怎么解决呢?调高灵敏度吗?
本人最近在用P/ACE MDQ(用的是未修饰过的毛细管柱)做超氧化物歧化酶(CU/ZnSOD)的含量测定,但是标准品的浓度达到1000ppm峰还是很小,用的缓冲液是pH5.36的磷酸缓冲液,内加SDS,有人做过SOD吗?请提供点意见!!!
日前,WTO秘书处发布了中国关于化妆品用二氧化钛标准草案的通报。该标准草案规定了化妆品用二氧化钛的分类、要求、试验方法、检验规则、标志、标签、包装、运输和贮存,标准适用于化妆品用二氧化钛粉体。该产品用于化妆品中主要起遮盖、改善肤色、增白及屏蔽紫外线的作用。
按工作场所标准160一步步来做五氧化二磷 每次都做不好曲线。因为160很多标准实在坑爹。所以论坛里查了大量的资料,大家也同样碰到了很多困惑,后面去查文献,改进的方法很多,但是真正的问题没解决。160里面 试剂配制写的很模糊,最后也没说要定容,注意事项里说氯化亚锡显色要一定的酸度,但是老标准是采用c(1/2H2SO4)来表示,到了新标准c(H2SO4) 居然数字都没变。开始我一直以标准上提到的标准的酸度来控制反应,算算是不用定容到10mL的。结果每次都做不好。 160坑爹的地方还表现在,配置氯化亚锡的时候也没提到要加热或者90度水预,直接溶到甘油里有点困难的,而且也不好滴加到管子里。后面我到门诊要了几根一次性吸尿样的吸管总算是加起来方便多了,如果不定容,氯化亚锡最好都要加到溶液里,沾到管壁上反应肯定成问题,但是如果定容到10mL就无所谓,而且定容不定容标准管吸光度是一样的,所以定容到10mL反应重复性好,而且更稳定,线性也好了很多,3个9一点问题没有。其实就是定容至10mL问题,160的标准还不知道有多少坑爹的地方。
大家好,我现在在用agilent 6890N、FPD检测器、DB-1701毛细管柱做有机磷类农药,总共需要做十一种农药,现在的问题是别的十种农药做出来都没问题,峰形,响应什么的都很好,只是氧化乐果做的时候,出5、6个峰,一开始以为是标准品污染,重配了0.3ppm的氧化乐果,并且清洗了进样针后,做出来任然是5、6个峰,想请教大家可能会是什么原因,谢谢。我用的是NY/T 761-2008中的单进样器单柱方法。
今年4月1日,我国向世贸组织技术性贸易壁垒委员会通报了国家质检总局和国家标准化管理委员会联合发布的化妆品用二氧化钛中国国家标准(编号ICS:71.060.20)规范文件。 该标准由中国石油和化学工业协会提出,中国化学标准化技术委员会无机化工分会(SAC/TC63/SC1)归口,上海、天津、江苏及河南四家化工企业共同起草。 标准规定了化妆品用二氧化钛的分类、要求、试验方法、检验规则、标志、标签、包装、运输和贮存,适用用于化妆品生产制造的二氧化钛粉体,该物品在化妆品中主要起遮盖、改善肤色、增白及屏蔽紫外线的作用。标准第5章涉及的重金属、砷、铅、汞四项指标以及第8章标志、标签要求为强制性,其他为推荐性要求。 化妆品用二氧化钛应分别符合表1 和表2 要求:表1 I类产品的要求项目指标锐钛型(A)金红石型(R)二氧化钛(TiO2)w/% ≥9898干燥减量 w/% ≤0.50.5灼烧矢量 w/% ≤0.50.5水溶物 w/% ≤0.50.3重金属(以Pb计)w/% ≤0.00200.0020砷(As)w/% ≤0.00050.0005铅(Pb)w/% ≤0.00100.0010汞(Hg)w/% ≤0.00010.0001pH 6.5~8.56~8白度(Wh) ≥9090细度(45 μm) ≤0.10.1
工业氧化铁和四氧化三铁的行业标准?如果谁有?请贴出来好吗?[QUOTE][COLOR=RED]请发贴前自己搜索一下标准号和名称...,否则不予应助!![/COLOR][/QUOTE]
食品安全国家标准 食品营养强化剂 氧化锌
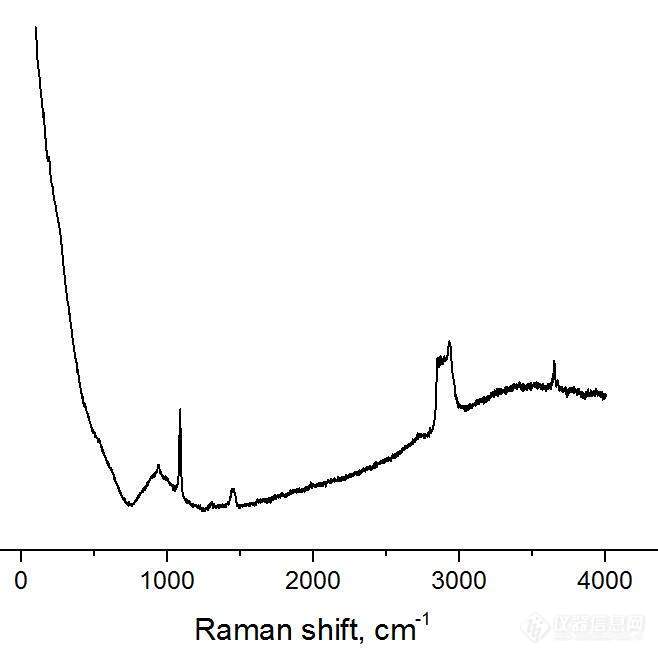
[color=#444444]拉曼光谱标定氧化物,碳化物 (如: 镁和锂的氧化物和碳化物,下图是我刚刚做的氧化表面的拉曼光谱)。如何查到金属氧化物,碳化物的拉曼特征峰位?有没有计算,推断方法;或者标准图谱可以查询?跪求大神解答~~[/color][color=#444444][img=,658,655]https://ng1.17img.cn/bbsfiles/images/2019/08/201908301108133406_1068_1701336_3.jpg!w658x655.jpg[/img][/color]
我用的是岛津2010GC, FPD检测器,DB-17柱子,仪器条件:进样口220度,柱箱起始温度130度,升温程序:130度保持2分钟,以6度/分升温到230度,保持2分钟.检测器250度.分别进0.1浓度的甲胺磷和氧化乐果单标都不出峰.只能连续进十几针高浓度的单标后才出峰,是什么原因?这样做会不会影响定量结果?请各位高手们帮解释一下.谢谢~
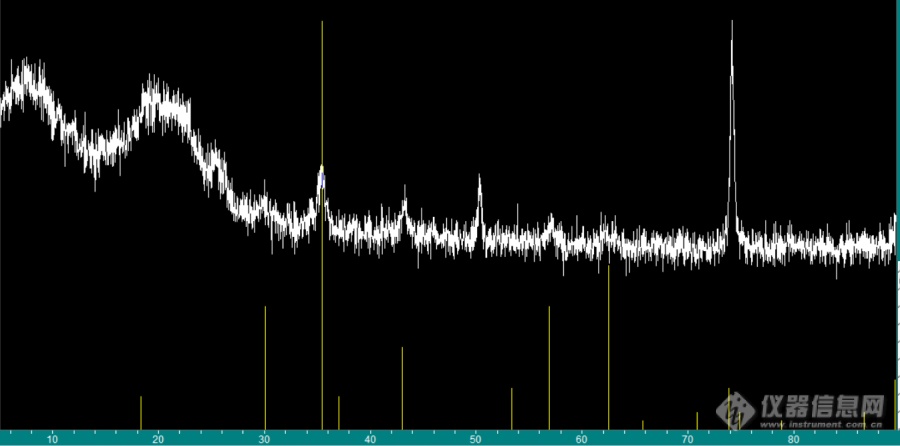
[img=,690,341]https://ng1.17img.cn/bbsfiles/images/2023/06/202306011101064604_9722_6006202_3.png!w690x341.jpg[/img]在75左右的这个峰非常高,不太懂为什么,目前用的是PDF#19-0629这个卡片来对的四氧化三铁。求各位大佬帮助分析,不胜感激
上周用做农残检测发现检测氧化乐果标样不出峰,其他样品如乐果、三唑磷等正常出峰。加大浓度上机检测也不出峰。请问该从哪些方面排查问题???
Talin?索马甜提取自非洲竹竽(Thaumatococcus Daniellii),现已正式被批准列入中国食品国家标准,继Talin?索马甜成功进入欧洲、日本和韩国等多个市场后,我们将Talin?索马甜进而带入了庞大的中国市场。Talin?为Naturex索马甜产品之品牌凭借30余载之经验,Naturex可谓索马甜的全球引领者。Talin?为一种多功能食品添加剂,于食品饮料具有口感改良作用,同时可掩盖不良口感,此外亦可增强风味从而改善糖和盐的口感。索马甜采自于西非雨林地带,其果实称为西非竹竽(Thaumatococcus Daniellii),此果通过水提取可确保索马甜的百分百天然特质。纯度标准及分析方法的建立Naturex于2010年启动Talin?索马甜在中国的食品添加剂法规项目的申请工作,此次得到中国卫生部官方批准并公布,着实是对我们团队做出努力的最佳回报。中国法规中的纯度标准以及严格的分析检测方法皆基于Naturex所开发和使用的独有的测试方法。使用范围及市场机遇此次荣获批准,为我们中国食品饮料行业带来了绝佳的机遇。Talin?索马甜目前可使用范围为绝大多数饮料类,加工坚果与籽类,焙烤食品以及餐桌甜味剂等。Naturex预见在“低热量”饮料类中索马甜将大显神通,因其可使口感更圆润,甜味更丰满;对于餐桌甜味剂,由于Talin?索马甜为一天然甜蛋白,加之其热量极低,可谓是更健康的选择。
五氧化二磷钼酸铵分光光度法 GBZ/T 160.30-2004(改进)一、 原理空气中的五氧化二磷或三氯化磷用吸收液采集,生成的磷酸与钼酸铵和氯化亚锡反应生成磷钼蓝,在680nm 波长下测量吸光度,进行定量。二、 仪器1、 多孔玻板吸收管。2、 空气采样器,流量0~3L/min。3、 具塞试管,10ml。4、 恒温水浴。5、 分光光度计。三、 试剂 实验用水为去离子水。1、 硫酸,ρ20=1.84g/ml2、 硫酸溶液,5mol/L:28.8ml 硫酸慢慢注入水中,定容至100ml。3、 吸收液:水。4、 氯化亚锡溶液:溶解2.5g 氯化亚锡于100ml 丙三醇中,室温下可使用1个月。(注:配置氯化亚锡溶液时要加热溶解或者置于90℃中水浴中溶解。)5、 钼酸铵溶液,50g/L。(注:应该采用四水合钼酸铵。)6、 五氧化二磷标准溶液:准确称取0.2454g 干燥过的磷酸氢二钾(K2HPO4),溶于水中,定量转移入1000ml 容量瓶中,再稀释至刻度,此溶液为100*g/ml 标准贮备液。临用前,用水稀释成10.0*g/ml 五氧化二磷标准溶液。或用国家认可的标准溶液配制。四、分析步骤1、 对照试验:将装有10.0ml 吸收液的多孔玻板吸收管带至采样点,除不连接空气采样器采集空气样品外,其余操作同样品,作为样品的空白对照。2、 样品处理五氧化二磷样品的处理:用采过样的吸收液洗涤吸收管进气管内壁3 次,将吸收液倒入具塞比色管中,摇匀。于沸水浴中加热15min,取出冷却。吸取5.0ml 放入另一具塞比色管中,供测定。若样品液中五氧化二磷浓度超过测定范围,可用吸收液稀释后测定,计算时乘以稀释倍数。3、五氧化二磷标准曲线的绘制:按表1配置标准管。向各标准管中加入0.5ml 硫酸溶液,摇匀;加0.2ml 钼酸铵溶液,混匀;加1 滴氯化亚锡溶液,摇匀;(应该定容至10mL,否则显色重现性很差)放置15min。以0标准管为参比,于680nm 波长(1cm比色皿)下测量吸光度,以五氧化二磷含量(ug)对相应吸光度绘制标准曲线。 表1 P2O5标准管 管号 10 ug/mL P2O5(mL) 超纯水(mL) P2O5含量(ug)1 0.00 5.00 02 0.20 4.80 23 0.40 4.60 44 0.60 4.40 65 0.80 4.20 86 1.00 4.00 10 五、结果讨论1、标准上说磷钼络合物还原成磷钼蓝必须在一定的酸度下进行,酸度过低则空白管呈蓝色。以氯化亚锡为还原剂时,最适宜的硫酸溶液浓度为0.80~0.95mol/L,以前采用的是;加入的量应该一致。显色达到稳定后,应尽快测定。实验证明定容至10mL,硫酸浓度降低并不影响显色和吸光度。反而是使显色更快更完全。2、 改进后的标准曲线直接安装标准来做无法做出标准曲线,经过改进后按表1测定五氧化二磷标准曲线,线性关系r=0.9997回归方程Y=0.004700+0.027200X 式中X为五氧化二磷含量,Y为五氧化二磷的吸光度值。3、改进后的方法的精密度和最低检出限用2、4、8 ug的五氧化二磷标准分别平行测定6次吸光度A值,RSD在3.70%~7.6%之间。另做10次0管样计算出10次中最低0零管A值对应的五氧化二磷含量,得出五氧化二磷最低检出限为0.2ug/mL。结果显示改进后的方法的精密度及最低检出限都满足比色分析的需要。完全按标准做无法得到满意结果4、改进后的方法准确度取一样品含量的五氧化二磷本底值,将溶液均分为9份,每3份一组。分别加入低、中、高3种浓度的五氧化二磷标准溶液,计算回收率。结果见表2.,证实改进的方法准确性是可靠的。 表2改进法测定 P2O5回收率(n=3) 样品 P2O5 含量(ug) P2O5 (ug) 平均回收率 (%) 加入量 测得量 1 5.13 2.00 2.10 1052 5.13 4.00 3.87 96.83 5.13 10.00 9.89 98.9 六、小结GBZ/T 160.30-2004只是GBZ/T 160标准中的相对有代表性的一个,其他标准很多地方也同样存在表述不详细甚至模棱两可的地方,需要我们自己做实验去确证和改进。这个实验关键就是最后定容至10mL,一旦这么做了,这个实验重复性不好,线性不好,准确性不可靠就迎刃而解了。

用DB1701(30*0.25*0.25)做氧化乐果、二唪农,两峰能分离,出峰时间相差0.13min,该柱子用两年后,柱效逐渐降低,同样的条件件,氧化乐果、二唪农完全重合,有无别的型号的色谱柱做这两种农药,分离效果较好的?http://ng1.17img.cn/bbsfiles/images/2013/05/201305231631_441244_1645480_3.jpg2011年氧化乐果、二唪农出峰情况。http://ng1.17img.cn/bbsfiles/images/2013/05/201305231635_441247_1645480_3.jpg2013年氧化乐果、二唪农出峰情况。
自从做了行标小版主以来,也没在论坛上为大家做点贡献,这次整理了一个大礼包--HG化工标准69种工业品打包上传(酸碱盐及氧化物)希望对大家有用处!觉得有用的请顶一下!我会再接再励的!谢谢大家[em62] [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=31606]HG化工标准69种工业品(酸碱盐及氧化物)[/url]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=35357]GBT 8753.1-2005 铝及铝合金阳极氧化氧化膜封孔质量的评定方法 第1部分 无硝酸预浸的磷铬酸法[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=35358]GBT 8753.2-2005 铝及铝合金阳极氧化氧化膜封孔质量的评定方法 第2部分 硝酸预浸的磷铬酸法[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=35359]GBT 8753.3-2005 铝及铝合金阳极氧化氧化膜封孔质量的评定方法 第3部分 导纳法[/url]
我做农田氧化亚氮,色谱柱采用PQ柱*2m。ECD检测器,六通阀进样,标准品进样情况很好,里边没有氧气和水,但样品进去直接出不来啊,直接掩盖了,还有就是标样出风在0.8分钟左右,是不是太早了啊第一次用色谱,求助啊http://simg.instrument.com.cn/bbs/images/default/em09509.gif
鄙人近日对氧化锆粉体做了表里改性,但是去找不到标准谱图进行对比,因此希望得到给位“大虾”的帮助!氧化锆基本信息如下:二氧化锆:Zirconium dioxide 或 Zirconia立方相:cubic;四方相:tetragonal;单斜相:monoclinic不胜感激!急用!!
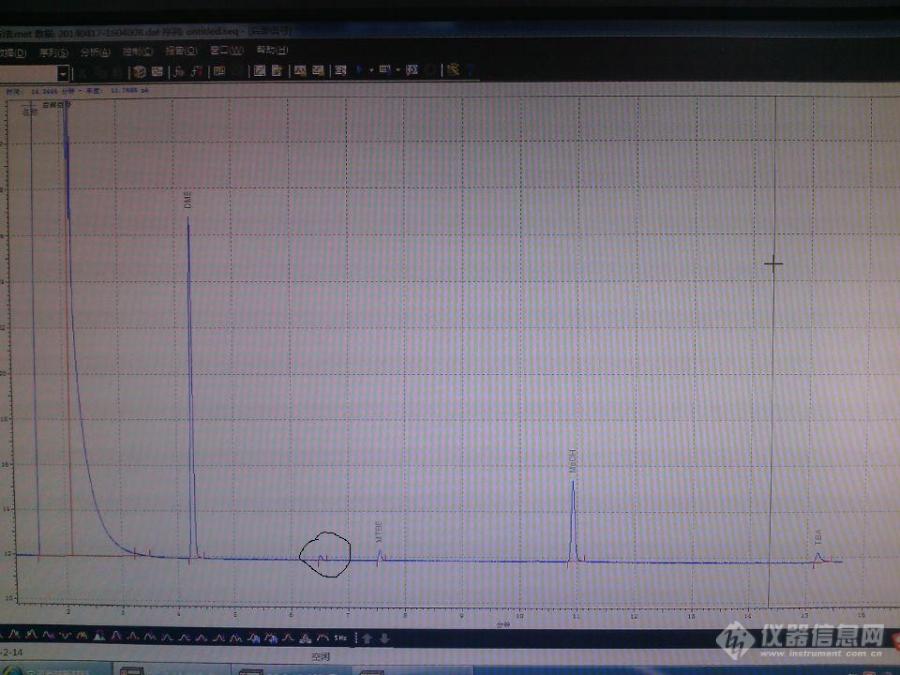
各位专家,我用的是GS-OxyPLOT柱子做的氧化物,在MTBE前面的组分峰是什么峰?分析的是混合碳四样品中的氧化物[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/09/201909061114480878_4156_1795523_3.jpg!w690x517.jpg[/img]
大家好! 想请教一个问题。调理食品(如裹有面包屑的未油炸的芋丸)要测其过氧化值,按标准应先前处理,前处理用石油醚进行浸泡过夜,隔天直接过滤浓缩,但浓缩完已经基本全干了,用三氯甲烷冰乙酸润洗浓缩瓶后进行滴定,结果过氧化值很高,(但是权威机构对同一样品的检测结果是未检出)不知道是什么原因啊?希望有人能指导一下。
大家有没有利用气相色谱法检测油脂类试样中的TBHQ,目前遇到问题如下:1、TBHQ标准试样采用气相色谱法检测出现双峰,两峰分离度超过2以上。2、经GC/MS确定保留时间短的目标物为TBHQ的氧化物,保留时间长的目标物为TBHQ,若只采用TBHQ制作标准曲线,线性关系较差,若将两目标峰合并制作标准曲线线性关系良好。因此怀疑采用GC方法检测,目标物TBHQ在进样口内被氧化,由于标准系列浓度不同,导致其氧化程度不一样,致使其采用单峰线性关系较差。3、不知道大家有没有采用GC方法检测该目标物的,有没有出现以上问题,若是没有能不能交流一下,谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP