南昌大学陈义旺团队在能源转换和存储领域取得重要研究进展
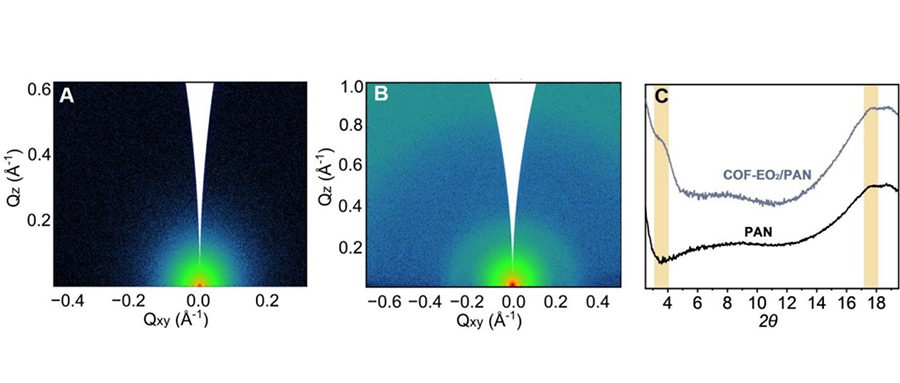
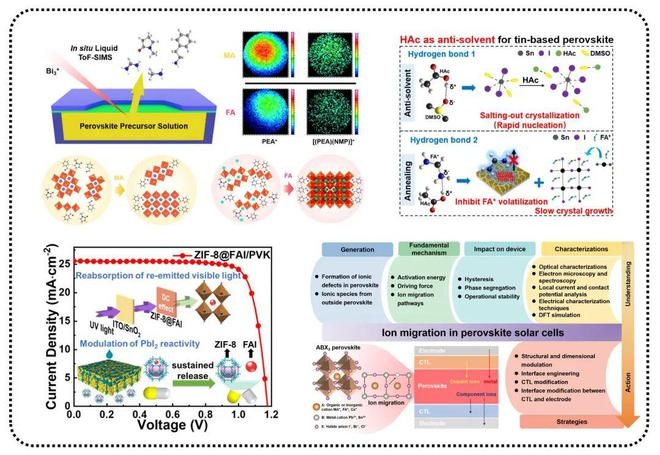
近日,南昌大学化学化工学院、高分子及能源化学研究院陈义旺教授团队在能源转化和存储领域取得重要研究进展。在能源转化领域,通过调节铅基/非铅基钙钛矿吸光层结晶行为,实现高效、稳定钙钛矿光伏器件。在能源存储领域,通过构造和调控多级纳米结构与电极界面,实现高效氧还原电催化剂和锌金属电池的制备。得益于简易的溶液加工方式、优异的半导体性能以及对柔性可穿戴设备的兼容性,钙钛矿太阳电池已成为光伏商业化应用中极具潜力的候选者之一。然而,相比于传统光伏技术长达20年的使用寿命,钙钛矿太阳电池的稳定性仍是制约其商业化应用的关键因素。作为制备钙钛矿太阳电池的初始材料,前驱体溶液中的高活性组分极易发生副反应,从而引发钙钛矿太阳电池的效率批次性以及稳定性问题。此外,由于对溶液表征手段的局限性,前驱体溶液中胶粒的组装行为对后续晶体的生长影响仍未可知。鉴于此,陈义旺团队利用先进的液体飞行时间二次离子质谱仪作为“分子眼”评估前驱体物种差异,刨析前驱体溶液接触空气后的化学演变,直观揭示钙钛矿前驱体溶液老化本质。同时,结合氢键强度变化与离子团簇含量差异,可视化低维钙钛矿前驱体溶液中胶粒组装与量子阱演化之间的内在联系,实现低维钙钛矿模组可印刷性的突破。为进一步论证氢键作用在形核结晶过程的普适性,在无铅钙钛矿体系中揭示了以盐析结晶为主导的反溶剂机制和以氢键强弱为依据的挑选反溶剂的通用规则,发展了以乙酸为代表的一类多功能绿色新型反溶剂。此外,针对两步法中碘化铅的残留问题,引入“多功能胶囊”概念构筑多孔通道,促进固液界面反应,并通过下转换效应提高光利用率,实现高效、稳定的钙钛矿太阳电池的制备。团队进一步总结了钙钛矿太阳电池中离子迁移的起源和抑制离子迁移的有效策略,并创新性地从整体器件的角度提出了抑制离子迁移的前瞻性方法,为开发高效、稳定的钙钛矿太阳电池提供了新思路。针对当前商业化的锂离子电池面临的性能、安全和成本等瓶颈,研发下一代环境友好型储能技术以提高器件功率密度、能量密度、安全性,降低制造成本显得尤为重要。得益于资源丰度高及绿色无污染等特性,水系锌基电化学储能器件极具发展前景。为提高水系锌空气电池的功率密度及稳定性,陈义旺团队通过在邻苯二甲腈功能化石墨烯表面经微波聚合原位生长铁酞菁聚合物,通过液相原位电荷剥离策略,制备得到铁酞菁聚合物纳米片纵向接枝于石墨烯的多级次纳米片,作为高效氧还原电催化剂用于液态和柔性准固态锌空气电池。此外,为克服锌金属负极面临的枝晶生长、腐蚀、钝化等问题,团队采用聚阳离子电解质-聚二烯丙基二甲基氯化铵(PDD)作为添加剂双向调控电解液和锌/电解液界面电场,改善Zn2+迁移行为,诱导Zn(002)优势沉积,成功构筑高可逆和高稳定性的锌金属电池。团队长期围绕能量转换与存储器件关键材料与技术等方面开展研究,发展了一套可全自动化印刷制备工艺,实现大面积柔性固态能量转换与存储器件(太阳电池、超级电容器、金属-空气电池)的制备、集成及应用,在专利技术和工艺优化中取得连续突破,为进一步的产业化提供了支撑。团队最新研究成果近期连续在化学和材料顶级期刊Angewandte Chemie International Edition,Advanced Materials和Energy & Environmental Science上发表(Angew. Chem. Int. Ed., 2023, 62, e2022157 Angew. Chem. Int. Ed.,2023, e202303177 Angew. Chem. Int. Ed., 2023, 62, e2023016 Angew. Chem. Int. Ed., 2023, e202302701 Adver. Mater., 2023, 2301852 Adver. Mater.,2023, 2302552 Energy Environ. Sci.,2023, 10.1039/D3EE00202K),南昌大学为论文第一及通讯作者单位。




























 我要推广仪器
我要推广仪器
 下载APP
下载APP