最近,接到一个项目,用氨基柱做糖酯的分析,样品里面含有吡啶、甲苯、叔戊醇等溶剂。这些溶剂有没有好的办法除去,而不损失糖醇、糖酯。若不除去,直接进液相色谱,会不会对柱子有损伤。
公司用的原料有叔戊醇,纯度要求大于等于百分之九十九,请问谁知道检测叔戊醇有国家标准吗???
测白酒里的异戊醇,出峰的时候后边一直有一个小峰,加标回收后小峰面积基本不变,但是加标后异戊醇峰甚至会变得更小,加标回收率也一直不稳定,有没有大佬帮帮我,太难了??。
我用DB-624(125-1334)测定乙醇中的异戊醇标准溶液,怎么还有一个峰和异戊醇连在一起呀,求如何分离?进样口:250,检测器FID:300,柱温:40,保持5分钟,40-260保持1分钟,以10/分钟。氮气流量30cm/sec
我是按5009做的,单点,结果已经做出来,于是上来参与讨论一下,发现自己做的杂醇油总量要比别人做的小,百思不得其解。后来看到有网友问异戊醇的出峰问题,好些人说只出一个峰,但我历来做的都是两个峰的,异戊醇旁边的峰是活性戊醇。以前用30m的柱子这两个峰是分不开,用CPWAX 57cb 50m的才行,这个柱子的是CNAS专家建议采购的,因为几年前CNAS现场评审考核时候就是因为异戊醇的问题,没有做好。所以现在我看到我做的数比别人的要小,但是不知道怎么报,虽然自己的数是可能对的,但如果有好多数人的结果都是把活性戊醇和异戊醇当成了异戊醇,那我的结果最后肯定是偏离值了。我试了一下,把活性戊醇和异戊醇当一个峰计算,那结果就和其他人一致。 A / B甲醇:0.110 / 0.0944杂醇油:0.198 / 0.180
新手求助,在进行杂醇油出峰时间确定的时候发现异戊醇的峰是3个峰连一起的这是正常的情况还是有杂质干扰。
标准上说异戊醇要加试剂后弃去水相,但异戊醇溶于水啊,没有水相可弃。是先将异戊醇和四氯化碳合并后再加试剂,还是异戊醇单独先加试剂,弃去水相再和四氯化碳合并?
哪位知道国产的异丁醇,异戊醇标准品哪有?
想要分离甲苯和正戊醇该用什么柱子?
安捷伦的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url] 柱子HP-5MS请问3-戊醇(沸点114-115),3-戊酮(沸点101.5)该怎么分离(3-戊酮含量较少),谢谢
自动进样,1微升,分流比20:1进样口温度230℃柱流速1ml/min柱温见图柱子 HP-INNOWAX检测器FID,250℃可以看下图,只有甲醇和乙醇有峰。异戊醇和异丁醇都没,之前,单跑异丁醇在2.9min,异戊醇在3.7min浓度都在10mg/ml。如何解决呢?今天又跑了一下,结果有4个峰,但是突然断电后,重新又跑了,就只有两个了,费解啊。http://ng1.17img.cn/bbsfiles/images/2015/01/201501071927_531517_2287675_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/01/201501071927_531518_2287675_3.jpg
[em0812]我新买回PEG-20M色谱填充柱(3m*3mm)老化后,进正丁醇样后发现正丁醇与异戊醇很难分离,有时还被正丁醇峰覆盖了,我选择的条件是:柱温110,进样器180,检测器180。后来我又改变了条件:柱温(80-110)、进样器(145-180),又调整载气(N2),还是分离不出异戊醇,各为高手有何解决办法?[em0808][em0810][em0811]
DNP混合柱,原来一直使用正常,最近突然出现内标与异戊醇无法分离的现象,可是其他的峰都正常,不知道什么原因?
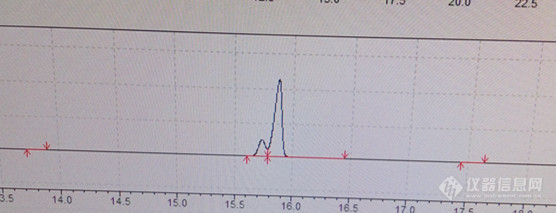
最近做伏特加中高级醇含量,用岛津GC2010-plus,安捷伦CP-WAX 57 CB毛细管柱,结果异戊醇标准品出了两个峰,哪位大神知道这是为什么啊?条件:进样口220,检测器250流速0.5 40度保留3min,5度每分钟升到100度保留10min,5度每分钟升到200度,保留5min[img=,556,213]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221337042752_4406_1850285_3.jpg!w556x213.jpg[/img]
实验室要分离 丙酮 异丙醇 异丙醚 戊醇 水还有一些醇类杂志求推荐柱子我自己查了一下用安捷伦 115-2132 不知道行不行通常选柱子有啥依据没有啊谢谢!!!
哪可买到色谱纯正丁醇,异丁醇,异戊醇,2-甲基丁醇,正丙醇标样?
哪可买到色谱纯正丁醇,异丁醇,异戊醇,2-甲基丁醇,正丙醇标样?
我用的东西电子的GC4009[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测白酒中的异戊醇总是出现拖尾的问题,柱温设过160℃、170℃和190℃,汽化室温度设过190和230℃,检测器温度也是设过190和230℃,每次都出现拖尾,积分的时候总把一个峰分成两个,真是头疼。请教各位老师,怎么解决这个拖尾的问题?
五种混标用二硫化碳溶解,毛细管柱为安捷伦DB1701,进样口温度250,检测器250,柱温35,程序升温至150,结果只检出甲醇,正丁醇,异戊醇。而乙二醇,异丙醇未检出,是我的仪器条件设置问题吗?求教!
请问,测定DMAC中的DMF和HAc可不可以用戊醇做内标物啊,如果不能,哪该用什么才好?谢谢!
用国家标准做水产品中的组胺,正戊醇提取时无法分离有机相,why?有何窍门?
环氧七氯、顺式环氧七氯、反式环七氧氯是3个不同物质吗?还是环氧七氯分为顺式环氧七氯与反式环氧七氯?但环氧七氯只有1个色谱峰哦??
国标中规定杂醇油由异丁醇和异戊醇合计,是分别将其计算后相加呢?还是将两者的峰面积或峰高相加,再算出来呢?
我在GDX-102气象色谱柱(热导池法)上测混合物(含乙酸。乙醇。正丙醇。异丁醇。异戊醇。乙酸乙酯。乙酸正丙酯。乙酸异丁酯。乙酸异戊酯),请问专家:怎样控制气象色谱的操作条件能把这些混合物各自的含量测出来,而且峰形较好
求助大家一个问题,顺式环辛烯在紫外光后可以转化为逆式环辛烯,我需要测定溶液中顺式环辛烯和逆式环辛烯的浓度,但是市面上只搜到了顺-环辛烯,我怎么样才能确定逆式环辛烯的浓度呢?谢谢大家解答~
做乙醇挥发性杂质,按药典做供试品b的4-甲基-2戊醇峰面积始终小于杂质峰,新人求指导
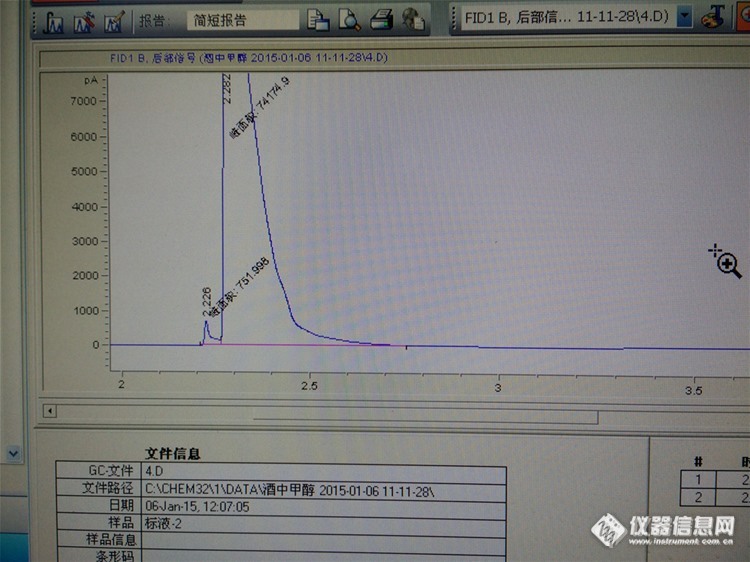
【问题】有做酒精中甲醇的吗?最近甲醇峰与叔戊醇内标峰,柱效都下降的快,峰又肥又矮,试过换衬管,老化柱,换柱,均没效(innowax的柱子,fid测其他物质没毛病)【回复】调下尾吹,氮气、氢氢和氧气流量比例要最佳,如果不行,在调节下分流比,调大点试试
氨基柱是同时可以用于正相条件和反相条件的,但是要注意到的是:正相溶剂和反相溶剂往往是不互溶的,对这一点的忽略可能会带给使用者一些麻烦。对于新购买到的柱子,首先请注意打开分析测试说明书,了解柱子的保存溶剂。如果保存溶剂与你将要使用的流动相极性不同不会互溶,请先用异丙醇过渡。 过渡过程中注意因异丙醇粘度较大,会导致柱压很高,适当调低流速即可。如果要使用的流动相中还含有缓冲盐类,建议在用分析流动相之前,先用不含缓冲盐的同比例流动相过渡,这样可避免缓冲盐在分析柱内的析出。☆ 柱效检测:这是判断一个用户是否有经验的指标之一,是用户是否习惯配制一些柱效检测标准溶液。说到柱效检测,可以向大家提供一个方法,而且很通用,你也可以用同样的标准溶液和流动相来检测硅胶柱和氰基柱。流动相:10%乙酸乙酯+90%正己烷流速1.0标准溶液:乙苯(如乙苯找不到就用甲苯代替呗)+苯甲酸甲酯检测波长254nm这个方法特别适用于新购柱子(新购柱子往往保存在正相溶剂中)时即进行检测,也适合在柱子在准备放置相当长一段时间之前进行充分清洗后进行检测作为记录留档。在平常的使用过程中,如果分析物是固定重复的,我们当然可以从对分析物的分离分析情况来作出判断;但如果分析物和分析条件不同时,定期检测柱效可以避免柱效下降导致分离不佳再分析查找原因这样浪费人力物力的过程。不过特别要注意的是,当氨基柱(或氰基柱)在反相条件中使用时,如用该条件检测,请注意流动相的换相过渡问题。 ☆ 氨基柱的使用:需要注意的是,氨基柱的键合官能团氨丙基要比C18,C8柱的键合官能团C18,C8要容易水解,所以首先要做好其使用寿命稍逊的心理准备,特别是当你的使用条件是反相条件下时。反相条件下使用时,要特别注意控制PH值范围,PH值越低越有发生水解的危险,流动相中水的比例越高当然也越有发生水解的危险。所以,在使用后以及准备长时间放置该柱时,必要的清洗和将氨基柱保存于纯的有机溶剂中是很好的保养措施。有一种情况是,当使用氨基柱进行酸性物质如果汁的分析时(分析其中的糖份),酸性物质的存在意味着质子的存在,可能会使略带负电荷的氨基官能团质子化,导致使用一段时间后对于某些类的分析物保留性质有所改变或表现在柱效下降。这时,Kromasil专家所给的建议是:用5-10倍的柱体积的含0.5-1.0%NH3的50-50乙腈-水溶液冲洗该柱(冲洗后当然要再用不含碱的流动相洗去多余氨),之后再进行分析这类酸性分析物时建议在流动相中略微添加少许氨如0.1%。☆ 氨基柱的清洗:简单说起来,正相条件下使用的氨基柱你就参照硅胶柱的清洗方法;反相条件下使用的氨基柱你就参照C18的清洗方法。※ 平时在正相条件下使用:首先用50倍柱体积的异丙醇清洗,因异丙醇粘度较大可适当放低流速;之后,用50倍的甲醇清洗;之后,用异丙醇过渡回到平常使用的正相流动相,即可。※ 平时在反相条件下使用:缓冲盐应及时冲洗,以及不能直接用纯甲醇冲洗缓冲盐等,属常识就不作特别交待了。用50倍纯甲醇冲洗;之后,用异丙醇过渡后,用二氯甲烷冲洗色谱柱;之后,再用异丙醇过渡回来到甲醇条件下。这些清洗方法,主要是针对当样品中有杂质逐步吸附累积到填料上时的处理方法,色谱柱表现行为为诸如柱压增高柱效降低等。
现在应该说这一点是清楚的:氰戊菊酯其中一个峰一定会与顺式氰戊菊酯重合,无论采取什么分离方式。因为氰戊菊酯本身就包含顺式氰戊菊酯。这种说法应该没错吧?问题是,我现在就是需要计算同一个茶样中,氰戊菊酯和顺式氰戊菊酯2个项目的含量。氰戊菊酯的含量我一直有做,就是用氰戊菊酯的标曲来计算,每个浓度的氰戊菊酯标样都有2个峰,计算时用的是组校准,而非浓度合计。而我的疑问就是:不论是出几个峰,茶样中顺式氰戊菊酯的含量该如何计算?应该用顺式氰戊菊酯的标样作曲线,从而计算含量;而不应该是直接用氰戊菊酯的标曲计算,对吧?不好意思,脑袋愚笨,多有打扰!!!
顺式氰戊菊酯进GC-uECD应该出几个峰?我进了针顺式氰戊菊酯单标(标准物质中心购买),居然出来了两个峰,同时在下一针还出现了一个鬼峰~安捷伦7890A DB17-30M。图见附件。







 我要推广仪器
我要推广仪器
 下载APP
下载APP