有什么比较好的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]方法定量检测N,O-双(三甲基硅基)乙酰胺中乙酰胺。用什么试剂做溶剂能够溶解乙酰胺同时又不与N,O-双(三甲基硅基)乙酰胺反应呢?
对乙酰胺基酚原料检验时,用超声和不用超声对比检验,用超声的检测结果偏低。这是啥原因呢?
最近用hp-5的色谱柱分析N,N-二甲基乙酰胺和其他的一些残留溶剂,溶剂采用二甲亚砜。N,N-二甲基乙酰胺不出峰,不知道是不是因为二甲亚砜和N,N-二甲基乙酰胺重合了的缘故。对了我的色谱条件是顶空程序升温,40度保持10分钟,10度每分钟升温至210度。二甲亚砜出峰时间在11分钟,较大的拖尾溶剂峰,大家帮忙看看是什么情况?
我们做对氟苯甲酸检测,别人推荐用N,O-双(三甲基硅基)乙酰胺反应后测他们的衍生物,可是我搞不明白这个反应原理,不知道是不是可以反应彻底,请诸位高手帮忙讲解一下,谢谢!
[font=&][size=18px]N,N-二甲基乙酰胺又称乙酰基二甲胺、乙酰二甲胺,简称DMAC,是一种非质子高极性溶剂,有微氨气味,溶解力很强,可溶解的物质范围很广,能与水、芳香族化合物、酯、酮、醇、醚、苯和三氯甲烷等任意混溶,且能使化合物分子活化,因此广泛用作溶剂及催化剂。在溶剂方面作为高沸点、高闪点、热稳定性高、化学性稳定的溶剂,可用于聚丙烯腈的抽丝溶剂、合成树脂及天然树脂、甲酸乙烯酯、乙烯基吡啶等共聚物及芳烃羧酸的溶剂;在催化剂方面可用于尿素加热制氰尿酸、卤代烷与金属氰化物反应制腈、乙炔钠与卤代烷反应制烷基炔、有机卤化物与氰酸盐反应制异氰酸酯等过程。N,N-二甲基乙酰胺还可用作电解溶剂及摄影用成色剂的溶剂、脱油漆剂、有机合成原料、农药及医药原料。从C8馏分中分离苯乙烯的萃取蒸馏溶剂等。[/size][/font]
求二甲基乙酰胺气相测试方法
腈纶成分溶解定量分析时,一般用二甲基甲酰胺还是二甲基乙酰胺比较好?
求二甲基乙酰胺质量标准 HGT 4770-2012 有急用,最好是能够下载的,多谢各位啦……
最近在做气相分析来料溶剂(二甲基乙酰胺、DMAC)的纯度,我用正丁醇作为内标物,做了一系列的实验来验证DMAC的校正因子。我想问的是,做校正因子测试的时候,内标物的加入量是不是固定不变的?如果拿已知DMAC的校正因子是不是可以测任何浓度、只要含DMAC的 溶液?
[color=#DC143C][size=4][B][center]请问哪位同仁有“二甲基乙酰胺”的标准及检验操作规程呀。现在我急需这标准及检验操作规程,希望能慷慨解囊,不胜感激![/center][/B][/size][/color]
如题,简单信息论文专业:环境工程论文主题:二甲基乙酰胺 萃取 精馏 气液相平衡 回收 废液处理论文分类:X78论文形态:共 71 页 约 57,297 个字符 约 5.041 M内容那位在学校的方便帮我下载下。不胜感激!
如题,在2.9min时乙酸乙酯出峰,在17min左右N,N-二甲基乙酰胺出峰,出峰时间为10min,峰形特别不好,四分之一圆形,我用的是岛津-2010,进样口和检测器温度均为200度,柱温为70度,分流比为10,急求!!!谢谢各位前辈!!
RT:10版药典对乙酰氨基酚中的对氨基酚及有关物质、对氯苯乙酰胺检测有个朋友公司开始做这个项目,药典采用的是C8色谱柱朋友想买热电的柱子,不知道有没有同行采用热电的柱子做过?有的话帮忙发一张图谱看看,谢谢!QQ:342832185
如题,要检测一样品的n,n-二甲基乙酰胺的残留量。初步打算用DMSO做溶剂,DB-624的柱子,检测器是MS。不知道方法合适不合适?请各位版友不吝指教,谢谢!
目前我在做二甲基乙酰胺(DMAC)的残留溶剂,DMAC峰拖尾因子太大了,都到了2.4了,条件:溶剂是DMSO,进样口:220°;程序升温:80(5min)40°/min 升到160°保持5min,再以60°/min,升到220°保持5min;柱子:DB-WAX;检测器:FID;手动进样。仪器为岛津GC-2010。也试过DB-FFAP,DM-624柱子,但拖尾因子都差不多,哪位大哥有啥好方法,帮我出出主意,不胜感激!
求助紫外分光光度法测定水中N,N-二甲基乙酰胺 这篇文献,哪位有能共享以下么,非常感谢
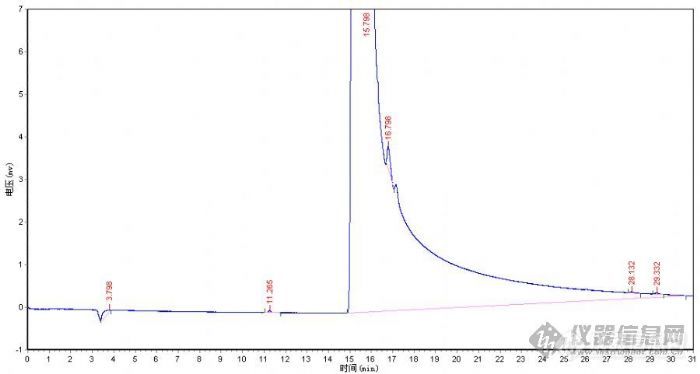
[em0702] 1 试样工业N,N-二甲基乙酰胺2 仪器2.1 福立GC9790[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]2.2 载气:高纯氮气2.3 检测器:FID3 色谱柱3.1 柱管:PEG-20M(30m/0.53mm/1.0μm)毛细管色谱柱4 色谱仪操作条件4.1 汽化室温度:220℃。4.2 检测室温度:250℃。4.3 柱箱温度:一段程序升温,以2℃/min速率从60℃升温至120℃维持2min 4.4 载气流速:6mL/min。4.5 氢气流量:0.148MPa4.6 空气流量:0.082MPa4.7 尾吹流速:40 mL/min5 进样5.1 进样方式:手动分流进样5.2 进样量:0.4μL二甲基甲酰胺(11.265min)、二甲基乙酰胺(15.798min)、醋酸(16.798min),其它峰还不能确定.我在上述条件下做,峰分离成这样子行吗?如果不行,我该如何修改操作条件?如何我要测定那些杂质的含量,我该用什么办法,怎么操作才好呢?我刚接触[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],什么都不懂啊,觉得好郁闷啊! 谢谢!!![img]http://ng1.17img.cn/bbsfiles/images/2007/11/200711262006_70988_1610765_3.jpg[/img][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=70989]图[/url]
请问含有DMA(N,N-二甲基乙酰胺)的反应液可以直接做LC监控吗??准备这样来做,取少许反应液,反应液里大部分DMA(DMA是溶剂),然后采用流动相稀释了,在进液相,不知道DMA会不会对柱子造成危害,C18柱子。同事们做监控是吧反应液吹干再稀释,但是DMA沸点156度,不好吹干啊。请问可不可以直接进液相,个人感觉问题不大啊。。反应前期用TLC监控,当TLC显示反应物没有的时候(可能是含量低看不到)再进LC。
正在做[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url] 实验,需要对样品进行衍生化,在买试剂的时候遇到一些问题。1、“N, O-双三氟乙酰胺BSTFA(含1%三甲基氯硅烷(TMCS))”这个试剂是液体还是固体呢?看文献描述应该是液体,但是搜出来的好像是固体。2、同一个试剂名称搜出来会有好几个规格,是不是随便选一个就行,只要名字一样3、不同试剂公司价格不一样该如何选择呢?
我在使用凝胶色谱仪时,采用DMAC(二甲基乙酰胺)为溶剂时,在25度下的聚苯乙烯的K和a是多少呀?
磺胺药物对氨基苯磺酰胺的合成目的原理Ar-NHCOCH3 + 2HOSO2Cl → p-ClO2S-Ar-NHCOCH3+ HClp-ClO2S-Ar-NHCOCH3 + NH3 → p-CH3CONH-Ar-SO2NH2 + HClp-CH3CONH-Ar-SO2NH2 + H2O → p-H2N-Ar-SO2NH2 + CH2CO2H仪器药品乙酰苯胺(自制) 5g(0.037mol);氯磺酸(d=1.77) 22.5g(12.5ml,0.19mol);浓氨水(28%,d=0.9) 35ml 浓盐酸,碳酸钠。过程步骤(1)对乙酰氨基苯碘酰氯在100ml干燥的锥形瓶中,加入5g干燥的乙酰苯胺,在石棉网上用小火加热熔化。瓶壁上若有少量水气凝结,应用干净的滤纸吸去。冷却使熔化物凝结成块。将锥形瓶置于冰浴中冷却后,迅速倒入12.5ml氯磺酸,立即塞上带有氯化氢导气管的塞子。反应很快发生,若反应过于激烈,可用冰水浴冷却。待反应缓和后,旋摇锥形瓶使固体全溶,然后再在温水浴中加热10~15min使反应完全。将反应瓶在冷水中充分冷却后,于通风中在充分搅拌下,将反应液慢慢倒入盛75g碎冰的烧杯,用少量冷水洗涤反应瓶,洗涤液倒入烧杯中。搅拌数分钟,并尽量将大块固体粉碎,使成颗粒小而均匀的白色固体。抽滤收集,用少量冷水洗涤,压干,立即进行下一步反应。(2)对乙酰氨基苯磺酰胺将上述粗产物移入烧杯中,在不断搅拌中慢慢加入17.5ml浓氨水(在通风橱内),立即发生放热反应并产生白色糊状物。加完后,继续搅拌15min,使反应完全。然后加入19ml水,在石棉网上用小火加热10~15min,并不断搅拌,以除去多余的氨,得到的混合物可直接用于下一步合成。(3)对氨基苯磺酰胺(磺胺)将上述反应物放入圆底烧瓶中,加入3.5ml浓盐酸,在石棉网上用小火加热回流0.5h。冷却后,应得一几乎澄清的溶液,若有固体析出,应继续加热,使反应完全。如溶液呈黄色,并有极少量固体存在时,需加入少量活性炭煮沸10min,过滤。将滤液转入大烧杯中,在搅拌下小心加入粉状碳酸钠至恰呈碱性(约4g)。在冰水浴中冷却,抽滤收集固体,用少量冰水洗涤,压干。粗产物用水重结晶(每克产物约须12ml水),产量3~4g。熔点161~162℃。纯品对氨基苯磺酰胺为白色针状结晶,熔点163~164℃。注意事项1.氯磺酸对皮肤和衣服有强烈的腐蚀性,暴露在空气中会冒出大量氯化氢气体,遇水会发生猛烈的放热反应,甚至爆炸,故取用时需加小心。反应中所用仪器及药品皆需十分干燥,含有氯磺酸的废液不可倒入水槽,而应倒入废液缸中。工业氯磺酸常呈棕黑色,使用前宜用磨口仪器蒸馏纯化,收集148~150℃的馏分。2.酰磺酸于乙酰苯胺的反应非常剧烈,将乙酰苯胺凝结成快状,可使反应缓和进行,当反应过于激烈时,应适当冷却。3.在氯磺化过程中,将有大量氯化氢气体放出。为避免污染室内空气,装置应严密,导气管的末端要与接受器内的水面接近,但不能插入水中,否则可能倒吸而引严重事故!4.加入速度必须缓慢,必须充分搅拌,以免局部过热而使对乙酰胺基苯磺酰胺水解。这是实验成功的关键。5.尽量洗去固体所夹杂和吸附的盐酸,否则产物在酸性介质中放置过久,会很快水解,因此在洗涤后,应尽量压干,且在1~2h内将它转变为磺胺类化合物。6.粗制的对氨基苯磺酰氯久置容易分解,甚至干燥后也不可避免。若要得到纯品,可将粗产物溶于温热的氯仿中,然后迅速转移到事先温热的分液漏斗中,分出氯仿层,在冰水浴中冷却后即可析出晶体。纯品对氨基苯磺酰氯的熔点为149℃。7.为了节省时间,这一步的粗产物可不必分出。若要得到产品,可在冰水浴中冷却,抽滤,用冰水洗涤,干燥即可。粗品用水重结晶,纯品熔点为219~220℃。8.对乙酰胺基苯磺酰胺在稀酸中水解成磺胺,后者又与过量的盐酸形成水溶性的盐酸盐,所以水解完成后,反应液冷却时应无晶体析出。由于水解前溶液中氨的含量不同,加3.5ml盐酸有时不够,因此,在回流至固体全部消失前,应测一下溶液的酸碱性,若酸性不够,应补加盐酸回流一段时间。9.用碳酸钠中和滤液中的盐酸时,有二氧化碳产生,故应控制加热速度并不断搅拌使其逸出。磺胺是一两性化合物,在过量的碱溶液中也易变成盐类而溶解。故中和操作必须仔细进行,以免降低产量。分析思考 1.为什么在氯磺化反应完成以后处理反应混合物时,必须移到通风橱中,且在充分搅拌下缓缓倒入碎冰中?若在未倒完前冰就化完了,是否应补加冰块?为什么?2.为什么苯胺要乙酰化后在氯磺化?直接氯磺化行吗?3 .如何理解对氨基苯磺酰氨是两性物质?试用反应式表示磺胺与稀酸和稀碱的作用。
哪位童鞋有《SN/T 3587-2013 进出口纺织品中N,N-二甲基甲酰胺和N,N-二甲基乙酰胺的测定 气质联用法》 这个标准,分享一下呗。谢谢啦http://simg.instrument.com.cn/bbs/images/brow/em61.gif
大家好! 本人在做氟乙酰胺衍生的时候,出现了一定问题,在这里请教各位朋友:在试管中加入氟乙酰胺的丙酮溶液与氢氧化钠固体。按理论来讲,氟乙酰胺与氢氧化钠反应生成氟乙酸钠和氨气,但是还怕引入水,我又加了一些无水硫酸钠。结果发现,试管中的盐(本来是白色)颜色逐渐变黄,越变越深。我没想清楚原因请教各位了?
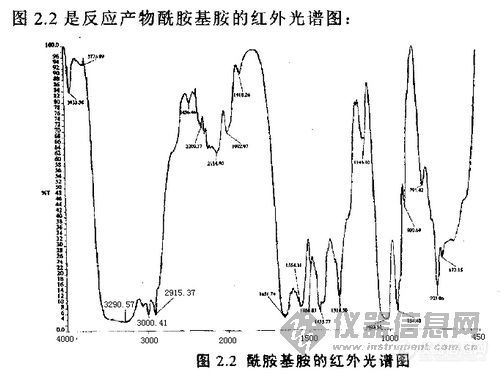
间苯二甲胺 和 己二酸反应生成 酰胺基胺,可是红外光谱上只能看见仲酰胺基的特征吸收峰,为什么看不到伯胺的峰?3000.41和2915.37处是不是伯胺吸收峰?是不是往后挪了?[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906080958_154498_1608859_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906080958_154497_1608859_3.jpg[/img]
在解红外光谱时常分不出来胺基(酰胺基)是与苯环相连的。还是在苯环侧链上。
如题,我做的一个单抗A用碘乙酰胺封闭二硫键,混合后产生了沉淀;用其他的单抗B样品就没事。而且其他的单抗样品B溶于单抗A的溶剂后再加碘乙酰胺也是澄清的。求问这是什么原因,有没有别的烷化剂或者别的合适的方法来封闭二硫键呢
顶空气相色谱法测定缬沙坦原料药中残留溶剂,以二甲基乙酰胺(DMA)作溶剂时,单独的对照不出峰,混合对照出峰,急求各位分析这是为什么?如何改进?顶空条件:DANI HSS86.50 顶空进样器,平衡温度100度,进样系统温度120度,传递管路温度120度,加压时间30秒,压力平衡时间5秒,注入时间30秒气相色谱条件:进样口温度200,检测器(FID)温度250,柱温:初始温度40,维持2分钟,以3度/秒升至100,再以30度/秒升温至200,维持2分钟;分流比10:1色谱柱:rtx-5ms,30m,0.25,0.251、以同样的样品(DMA溶解)手动进样出峰,但顶空不出峰。重复前期品种的方法,配置以水溶剂的甲醇、乙酸乙酯混合对照溶液,顶空进样,结果出峰,这是不是说明问题与DMA有关?2、DMA沸点166度,有没有可能是因为它在系统中冷凝?个人将顶空进样系统温度升至180度后,开始几针样品好了(但仍不确定是不是因为温度的原因),但过了一会单独的对照又不出峰了?3、因为中国药典标准中是用二甲基乙酰胺,所以用它了请各位帮忙分析一下,急死了!有什么没有说明白的地方,请指出,非常非常感谢!
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]测有机酸,色谱柱是DB-5MS,是非极性的图层,需要用到 N,O-双(三甲基硅基)三氟乙酰胺(1%三甲基氯硅烷),这个试剂含水吗?会损伤柱子吗?
我是从事有机合成分析工作的,试验中有一步氰基乙酰胺的含量对结果很重要,探索用液相建立起定量方法,尝试了甲醇-磷酸水溶液的流动相,试过C18和C8的柱子,其保留时间,总是在2分钟左右,请您赐教解决的办法,我用液相建立分析方法不是很熟练,也请您能多提意见,谢谢!
求乙酰胺的红外谱图!!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP