本人现在用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测硬脂酸镁,为什么对照品溶液中有很多杂峰,我用的正庚烷是色谱纯级别的,还有到最后计算是用不到对照品的浓度,那为什么还要进对照品?请各位老师指点
今天我把昨天的两份对照品储备液分别稀释成对照品溶液,顶空进样,但是第一份对照品溶液待测组分未出峰,第2份对照品溶液出峰正常,我想问下隔24小时以后再用待测组分都会挥发完全吗?为什么第二份正常出峰呀?对照品溶液里面的组分分别是乙醇,乙酸乙酯,丙酮,二氯甲烷,水为溶剂
大家经常做气相需要测对照品溶液,有时对照品溶液的出峰面积在不同时期可能差异较大,大家遇到过这样的问题吗?问题:取甲醇、乙酸乙酯、甲苯适量,精密称定,用DMF溶解稀释定容制成每1ml中约含甲醇60ug、乙酸乙酯100ug、甲苯20ug的混合溶液。那么我们实际称取的对照质量应该在什么范围内是可以接受的?因为称量的差异会导致对照品出峰面积的差异。问题:如何能用天平称准对照试剂的量?(有机溶剂甲醇、乙腈、二氯甲烷等)换算成体积量取还是直接称取?
作为质控的标液,是对照品还是算标准品?
在做国标 GB5009. 28 — 2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定对照品配制疑惑 标准溶液配制:苯甲酸、山梨酸和糖精钠(以糖精计)标准储备溶液( 1000mg / L ):分别准确称取苯甲酸钠、山梨酸钾和糖精钠 0.118g 、 0. 134g 和 0.117g (精确到 0.0001g ),用水溶解并分别定容至 100mL 。于 4℃贮存,保存期为 6 个月。当使用苯甲酸和山梨酸标准品时,需要用甲醇溶解并定容。红色这句话的意思是想用适量的甲醇溶解,然后在用水定容,还是用甲醇定容,感觉怪怪的?有那位老师可以指导一下, 共享一下经验,谢谢。
[size=3]不知大家注意没有,在2010年版药典中,特别是UV-Vis测含量,在“对照品溶液的制备”中,往往是准确指出精密称取的对照品的量,例如,2010年版药典一部第5页,人工牛黄中胆酸的含量测定项下,胆酸对照品溶液的制备:取胆酸对照品12.5mg,精密称定,置25ml量瓶中,加60%冰醋酸溶液使溶解,并稀释至刻度,摇匀,即得(每1ml中含胆酸0.5mg)。而,HPLC或GC等测含量,大多的表述是,如同是第5页,八角茴香中反式茴香脑的含量测定,对照品溶液的制备:取反式茴香脑对照品适量,精密称定,加乙醇制成每1ml含0.4mg的溶液,即得。这两种表述有何不同?[/size]
[color=#444444]我在按中国药典分析熊去氧胆酸片时,对照品的液相色谱峰有两个,4min左右和7min左右,峰高相近。但样品只有一个7min的峰,4min仅有一个很小的包。如果用对照品中7min的峰进行样品含量的计算,含量是合格的,请问对照里为什么有两个峰?已经排除是胆酸或鹅去氧胆酸了,而且用不同来源(中检所和某厂供)的对照品都是两个峰。[/color][color=#444444]谢谢各位![/color]
那里有丙烯酸对照品或标准品?如果找不到能否以自己精制过的样品当对照品?
近日在做一个日本上市缓释制剂的仿制,对其释放度检查标准有些疑问,特别是其供试品的吸收度计算方法很特别,希望园里的老师给分析指点一下,谢谢!释放度的测定取本品,照释放度测定法,采用溶出度测定法第二装置,以Ph1.2盐酸溶液500ml为溶剂,转速为每分钟100转,依法操作,经1小时时,取溶液10ml,过滤,作为供试品溶液(1);弃去上述各容器中的酸液,加已预热至37±0.5℃的Ph7.5磷酸盐缓冲液500ml,继续运转至2小时时,取溶液10ml,过滤,作为供试品溶液(2);并补加同体积的Ph7.5磷酸盐缓冲液,继续运转至5小时时,取溶液10ml,过滤,作为供试品溶液(3);另取对照品约0.1g,精密称定,置50ml量瓶中,加甲醇适量使溶解,并稀释至刻度,摇匀;精密量取此溶液各1ml,置100ml量瓶中,分别加Ph1.2的盐酸溶液和Ph7.5磷酸盐缓冲液稀释至此刻度,摇匀,作为对照品溶液(1)和对照品溶液(2)。照分光光度法,供试品溶液(1)在360nm和450nm波长处测定吸收度,计算吸收度差值;其他各对照品溶液及供试品溶液均在360nm波长处测定吸收度,分别计算不同时间的释放量。
近日在做一个日本上市缓释制剂的仿制,对其释放度检查标准有些疑问,特别是其供试品的吸收度计算方法很特别,希望园里的老师给分析指点一下,谢谢!释放度的测定取本品,照释放度测定法,采用溶出度测定法第二装置,以Ph1.2盐酸溶液500ml为溶剂,转速为每分钟100转,依法操作,经1小时时,取溶液10ml,过滤,作为供试品溶液(1);弃去上述各容器中的酸液,加已预热至37±0.5℃的Ph7.5磷酸盐缓冲液500ml,继续运转至2小时时,取溶液10ml,过滤,作为供试品溶液(2);并补加同体积的Ph7.5磷酸盐缓冲液,继续运转至5小时时,取溶液10ml,过滤,作为供试品溶液(3);另取对照品约0.1g,精密称定,置50ml量瓶中,加甲醇适量使溶解,并稀释至刻度,摇匀;精密量取此溶液各1ml,置100ml量瓶中,分别加Ph1.2的盐酸溶液和Ph7.5磷酸盐缓冲液稀释至此刻度,摇匀,作为对照品溶液(1)和对照品溶液(2)。照分光光度法,供试品溶液(1)在360nm和450nm波长处测定吸收度,计算吸收度差值;其他各对照品溶液及供试品溶液均在360nm波长处测定吸收度,分别计算不同时间的释放量。
对制品浓度要求0.3mg/ml第一个问题,算稀释倍数的时候,0.2ml算不算进去?粉末的对照品,是不算进去的,按1算!第二个问题,取样量(质量)怎么算?粉末的是用天平称,这种液体的对照品呢?怎么算?如果称的话,如果称?还是按密度算出质量?[img=,690,516]https://ng1.17img.cn/bbsfiles/images/2019/10/201910181106011657_3849_4008962_3.png[/img]
高效液相色谱仪对照品的纯度要求是多少?
目前我们实验室用的维生素A醋酸酯的对照品的供应商断货了,求问一下大家都用的是哪些供应商的对照品?我们也可以去买。我们试用过Sigma的和USP的发现都不行。Sigma的是实际含量和COA上的含量出入较大。USP的是一个混合物有全反式的和CIS的,由于我们不是用的中国药典附录上测定维生素A的方法,所以我们的液相分不开这2种物质,所以也不能用。
我按药典的配制方法:取碱式品红0.2g,加热水100ml溶解后,放冷,加亚硫酸钠溶液 (1→10)20ml、盐酸2ml,用水稀释至200ml,加活性炭0.1g,搅拌并迅速滤过,放置1小时以上,即得。 本液应临用新制。我配了好多次配出的都是黄色的。请教各路神仙,问题出在哪里?我用焦亚硫酸钠和无水硫酸钠都试过了,都不行。在加入亚硫酸钠溶液后一会儿就出现很多沉淀,加入盐酸后沉淀都消失了。此时产生的沉淀是什么?我配这个试剂是为了检测白蜂蜡的丙三醇与其他多元醇项目丙三醇和其它多元醇 取本品0.20g加氢氧化钾乙醇溶液(取氢氧化钾3g,加水5ml使溶解,加乙醇至100ml,摇匀,即得)10ml,加热回流30分钟,取出,加稀硫酸50ml,放冷,滤过,用稀硫酸洗涤容器和滤器,合并洗液和滤液,置同一100ml量瓶中,加稀硫酸稀释至刻度,摇匀,作为供试品溶液。取10ml纳氏比色管两支,甲管中精密加入供试品溶液1ml,加0.05mol/L高碘酸钠溶液0.5ml,混匀,放置5分钟,再加品红亚硫酸试液1.0ml,混匀,不应出现沉淀;然后将试管置40℃温水中。在水温下降过程中不断旋转试管,观察10~15分钟;乙管中精密加入0.001%丙三醇的稀硫酸溶液1ml,与甲管同时依法操作,甲管中显出的颜色与乙管比较,不得更深。(以丙三醇计,不得过0.5%)加入我配的品红亚硫酸钠样品盒对照就会出现沉淀。不知产生的是什么沉淀?问题出在何处?当我加两毫升品红亚硫酸溶液时沉淀消失,这又是为什么?我已经被这个实验困了好久了,请教大家,帮帮忙啊,感激不尽!
各位大神,请教一个小白的问题,关于HPLC中对照品的配制。外标法测定麦芽糖浓度。麦芽糖对照品是99%纯度的一水合物,请教问题是:真空干燥24h后,对照品中的一水合物的水是否会被去除掉,称量的时候是按一水合物的分子量计算来称量,还是按没有水的纯麦芽糖分子量来称量比如要求称1g就在天平称1g?
其实我想这么做的原因就是想节约点成本,因为做西青果药材的,对照品没食子酸用50%甲醇溶解,样品也是用50%甲醇溶解,地榆这个药材也是没食子酸,但是浓度稍微低点,我就想用做西青果的对照稀释一下就好,免得再次称对照,地榆药材是用水处理的,药典上写着是称没食子酸适量,用水溶解,现在里面有甲醇,会影响准确性吗?
药典 金钱草含量测定以甲醇一O.4%磷酸溶液(50:50)为流动相;检测波长为360nm,温度30℃。 对照品溶液的制备 取槲皮素对照品、山柰素对照品适量,精密称定,加80%甲醇制成每1ml各含槲皮素4μg、山柰素20μg的溶液,即得 供试品溶液的制备 取本品粉末(过三号筛)约1.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50ml,密塞,称定重量,加热回流1小时,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过。精密量取续滤液25ml,精密加入盐酸5ml,置90℃水浴中加热水解1小时,取出,迅速冷却,转移至50ml量瓶中,用80%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。 本品按干燥品计算,含槲皮素和山柰素的总量不得少于O.10%。
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。
求教:中药对照品的减压干燥如何做?我的做法是在减压干燥器内另放一装有少量五氧化二磷的蒸发皿,这样做对吗?但这样做在上高液时对照品峰都出现问题,要不是拖尾,要不是峰面积不稳定.请教!!!!!!!!!!![em53]
现在我们公司增加辅料乙醇和醋酸钠的红外检测,需要购买红外乙醇对照品和醋酸钠对照品。请各位大侠提供除了中检所外,能够在一个星期内买到的厂家。谢谢大家了!!!!
对照品浓度怎么算呢
液相色谱检测,产品主峰峰纯度达不到要求,购买的中检所和EP对照品主峰纯度也不行。
如何检测石油醚,乙酸甲酯和丙酮???石油醚本身就有几个峰,要做内标法找不到对照品。用异丙醇或正丙醇做内标物可以嘛?哪个好一些?DB-WAX柱子?进样口、柱温、检测器温度如何??谢谢
已知样品中含有醇类物质,如何使用高校液相色谱测试确定该醇类物质?对照品如何配制?
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
[size=3][color=#ff6600][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检薄荷桉油含片中薄荷脑的含量,仪器条件和操作方法如下[/color]:[/size][size=3][b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] 采用弹性石英毛细管柱(30m×0.32mm×0.25[/font][/size][font=宋体][size=3]um )(PEG)聚乙二醇为固定液,进样口温度220℃,检测器温度为250℃。分流进样。[/size][/font][font=宋体][size=3]程序升温,初始90℃,保持1分钟,每分钟5℃升至170℃。理论塔板数按薄荷脑峰计[/size][/font][font=宋体][size=3]算应不低于10000。薄荷脑与内标物质峰的分离度应大于4。[/size][/font][size=3][b][font=宋体]校正因子测定[/font][/b][font=宋体] [/font][font=宋体]取水杨酸甲酯适量,加丙酮稀释成每1ml含1.0mg的溶液,摇匀,作为内标溶液。另取薄荷脑对照品适量,加丙酮稀释成每1ml含1.0mg的溶液。精密取内标溶液与对照品溶液各5ml,置25ml量瓶中,加丙酮至刻度,摇匀,精密吸取1μl注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],计算效正因子。[/font][/size][size=3][b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]取本品20片,精密称定,研细,取粉末适量(约10片量),精密称定,置具塞瓶中,精密加入丙酮25ml,振摇30分钟,滤过,精密量取续滤液10ml与内标溶液5ml,置25ml量瓶中,加丙酮至刻度,摇匀,精密吸取1μl注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测定,计算,即得。我按上述方法操作,用的机子是岛津GC-14C,柱子是0.53×30的PEG-20M,各气体流量也正常,但在做时发现只有水杨酸甲酯的内标峰出来,保留时间大约在7分钟的样子,而进薄荷脑对照品怎么没有薄荷脑的峰呢?进样品同样没有薄荷脑的峰,只有内标峰,请大家帮帮忙分析一下原因出在哪里?[/font][/size]
我的产品与对照品只少一个氨基酸,怎样用HPLC测含量?求教高人指点。
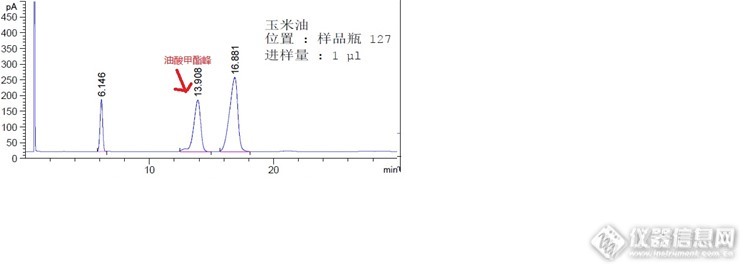
最近做了一个中药材中的油酸含量测定,用的是衍生后上气相的方法测定。采购先是从成都的一个卖参照品和中检所对照品的中介购买了一瓶油酸参照品(纯度有99%)。谁知......按药典方法做下去,这个参照品除了试剂峰竟然没有主峰出来,但样品是出峰正常的啊。后来我又找来了玉米油作参照,玉米油的棕榈酸油酸亚油酸硬酯酸4个大主峰都是有出峰的啊。通知采购联系卖家,卖家后来补寄了一瓶标示中检所出品的油酸对照品。很可惜的是,同时的制备操作,样品是出峰,那个中介补寄的标示中检所的油酸对照品一样是没主峰出来。这时,虽然我有10多年实验室经验,但主任一样http://ng1.17img.cn/bbsfiles/images/2014/09/201409271814_515972_1621232_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/09/201409271815_515973_1621232_3.jpg开始怀疑是我操作的问题,在没证据情况下,警告我,你说中检所的产品都有问题,我宁信中检所也不信自己手下了。不得已,我申请买了一瓶CP级的油酸甲酯作参照(因CP级的才30元,就算实验失败化费也不贵),这个CP级油酸甲酯很挺争气,真的出峰了。这时,主任消除了对我的怀疑,相信这个中介卖假货。下图是中检所油酸对照品和玉米油参照的色谱图。http://ng1.17img.cn/bbsfiles/images/2014/09/201409291648_516385_1621232_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/09/201409291648_516386_1621232_3.jpg大家都说说,你们买对照品是否也贪便宜,从中介那里拿货,其实,从中介那买对照品,也就是打个折扣而以,9折.....95折什么的。通过这次交易,明白了,不是中检所购买,很可能会买到山寨对照品.........
你们好,我想请教一下,用液相做氨基酸检测所需要用的标准品的纯度应该在多少以上啊?是不是用国家级标准物质会浪费?今天联系了很多供应商,想请购苏氨酸单独标准品,国家标准物质网上的苏氨酸对照品的价格在70元(100mg/瓶),标准物质750元/瓶(200mg/瓶),和别的供应商联系说中检所的标准品要600多元/瓶,纯度在99%,现在我就不确定了,到底标准品和标准物质是不是一样的,是不是检测的范围有所局限?请高人明示!谢谢
中检所提供的乙醇对照品乙醇浓度是多少?







 我要推广仪器
我要推广仪器
 下载APP
下载APP