苯乙酸是医药、农药、香料等有机合成的中间体。在医药工业中用于青霉素、地巴唑等药物的生产。苯乙酸经氯化、酯化得到α-氯代苯乙酸乙酯,用于稻丰散和乙基稻丰散的生产,这两种农药是广谱性有机磷杀虫剂。苯乙酸本身也是农药植物生长激素。苯乙酸广泛存在于葡萄、草莓、可可、绿茶、蜂蜜等中。苯乙酸在低浓度时具有甜蜂蜜味,在1ppm以下仍具有甜味,是一种重要的香料成分。苯乙酸还具有很强的杀菌作用。
求助各位同行:苯乙酸的国标或行标(HGB3444-62)全文.多谢了.我的邮箱:zjp9933@163.com
[color=#444444]甲酰基苯乙酸甲酯的色谱含量测试中。由于甲酰基苯乙酸甲酯会有醇酮互变性质,在液相色谱(乙腈:水=50:50)下出现三个峰。但是在LC——MS下从第一个峰开始到第三个峰这之间所有的时间都出现了M+1峰(包括峰之间的)。谁能告诉我这是怎么回事。[/color][color=#444444]有哪个大侠做过甲酰基苯乙酸甲酯的含量测试的,求指导。[/color]
目前所做项目需要液相检测联苯乙腈(原料)和联苯乙酸(产物),但两者出峰时间一致,已尝试多种方法进行混合样分离,但始终只有一个尖峰显示,没有任何分离趋势,目前试过的方法有:甲醇:0.05%磷酸水=70:30(C-18长柱);乙腈:水=70:30;乙腈:0.1%氨水水溶液=70:30(NX-C18);甲醇:0.1%醋酸水=55:45(C18短柱)。求助是否有可行的方法能够较好的分离两种物质。谢谢。
如题,在合成中有一步反应是用邻羟基苯乙酸制备其二钠盐,其中产物中可能含有的成分有邻羟基苯乙酸、邻羟基苯乙酸的一钠盐、二钠盐,请问如何建立检测方法将其分离呢?谢谢! 试过液相的方法,但是分不开,也试过双相滴定,但是里面还有过量的NaOH,影响结果,也试过用酚羟基的显色反应,但这个又太灵敏了,无法定量。请大家指导一下吧。
[color=#444444]求助一下啊!!!![/color][color=#444444]我做的羟基苯乙酸去送样,结果GC分析人员做出了两个峰,做了两次都是这样。[/color][color=#444444]我的样品应该是纯的,是分析方法不对,还是其它什么原因啊?有没有哪个遇到这个的情况呢?求助哈[/color]
[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#333333]4-羟基苯乙酸酯[/color][/size][/font],[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]CAS No.:[b]58556-55-1[/b][/color][/size][/font][font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]分子式:C[sub]1[/sub][sub]0[/sub]H[sub]1[/sub][sub]2[/sub]O[sub]3[/sub][/color][/size][/font]
[color=#d40a00][size=2]维权声明:本文为[font=Times New Roman]11093661[/font]原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[/size][/color] 青霉素发酵过程中利用液相分析对发酵液中的苯乙酸,效价检测存在着很是矛盾的主体,那就是发酵过程中的效价检测是批量检测,在检测过程中青霉素效价有高有低,这样就不利于药典中规定的标准品与待测品的含量大致相同的规定,这就不可能每做一个样品就做一个标准,这样不实际也不利于节约成本的。那么就需要我们做一个线性范围来使在实际检测的过程中的效价范围在线性范围类,这样就有利于测样的准确性。本人通过长期反复工作实践发现在青霉素发酵过程中发酵液的稀释倍数在100倍以上时,出现了这样一个情况:在检测效价的过程中对苯乙酸检测的结果很不稳定,特别是长期工作的液相更是如此,本人在工作中试验发现原因主要是基线的漂移造成了这个现象。而检测苯乙酸对生产发酵中的地位相当重要,苯乙酸是青霉素发酵过程中的主要原材料之一,而苯乙酸的多少又决定了发酵水平高低。所以说苯乙酸的检测也同样重要。 而效价,苯乙酸是同时检测出来的,如果稀释倍数大于100后解决了效价检测的准确性得到了提高,但是苯乙酸的检测准确性也就低了。所以这个矛盾主体也就出现了。本人在长期的检测中实践发现如果分开来检测,也就是两次检测,而对于两次检测过程中的效价与苯乙酸两种物质稀释倍数采取不同的稀释倍数这样有利于检测结果的准确性。或者对苯乙酸检测采取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测同时稀释倍数根据苯乙酸残量的多少采取不同的稀释倍数这样有利于检测结果的准确性,重现性。而根据本人了解某特大型青霉素发酵公司也就是只是对效价采取的稀释倍数的不同,这样提高了效价的准确性而导致了苯乙酸残量的检测的准确性也就降低了,导致发酵水平无法与成都某特大型青霉素发酵公司的水平相比较。这就说明了苯乙酸检测尤为重于效价检测。而分两次检测或气,液相同时检测这样不利于成本降低,本人通过长期的摸索,比较发现利用苯乙酸的多少来决定稀释倍数后,再根据效价的高低采取不同的标准品的含量,这样就利于检测的准确性与成本的降低——相当于分段检测。 本人事先声明这个纯属个人自己摸索试验得出的结论,在实际生产,检测中虽然得到了应用,但由于自己的试验结果,水平有限所以具体过程没有完全介绍,十分抱歉。
完全按GB29708-2013的方法处理,衍生化后质谱检不出五氯苯乙酸酯,不知道哪里有问题!请做过这个项目的指点。
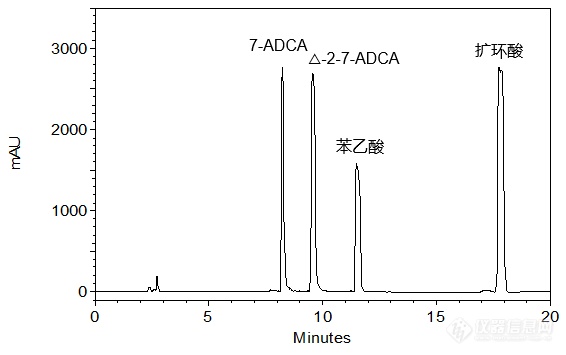
[align=center][b]4种头孢中间体的共同分析[/b][/align][align=right][b]——7-ADCA、△-2-7-ADCA、苯乙酸及扩环酸的分析[/b][/align][align=right][b][/b][/align]客户提供了7-ADCA(7-氨基去乙酰氧基头孢烷酸),△-2-7-ADCA、苯乙酸及扩环酸原料,希望本实验室依据客户所提供的色谱条件筛选合适的C[sub]18[/sub]色谱柱,实现以上4种化合物的稳定良好分析。本实验室参考客户提供的色谱条件,首先尝试使用经过聚合物包被处理的中等极性色谱柱——CAPCELL PAK C[sub]18[/sub] MGII对客户所提供样品进行分析,使用PDA检测器进行检测。如图1,在高浓度大体积进样的情况下,各色谱峰发生严重过载现象,出现平头峰;如图2,降低进样体积至5 μL可得到相对良好峰形,且各组分间能够得到良好分离,分离度均在10.0以上(结果详见表1)。[align=center][img=,566,355]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537444868_8925_2222981_3.png!w566x355.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,575,365]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545063985_5910_2222981_3.png!w575x365.jpg[/img][/align][align=center]图2 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表1 CAPCELL PAK C[sub]18[/sub] MGII分析结果详表(进样量:5 μL)[/align][align=center][img=,553,136]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537465105_7074_2222981_3.png!w553x136.jpg[/img][/align][align=center][/align][align=left][img=,579,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545307700_4011_2222981_3.png!w579x319.jpg[/img][/align][align=left][/align][align=left]为使客户有更多色谱柱选择,本实验室也尝试了能够在纯水系流动相下稳定使用的高极性色谱柱——CAPCELL PAK C[sub]18[/sub] AQ进行分析。如图3,几种头孢中间体的整体保留有所增强,而高浓度上样仍会出现与MGII色谱柱相似的过载现象;如图4,降低进样体积进行分析,可得到良好结果,同时发现扩环酸有一定程度的拖尾(见表2)。[/align][align=center][/align][align=center][img=,527,377]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546103944_1716_2222981_3.png!w527x377.jpg[/img][/align][align=center]图3 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,525,374]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546123571_3070_2222981_3.png!w525x374.jpg[/img][/align][align=center]图4 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表2 CAPCELL PAK C[sub]18 [/sub]AQ分析结果详表(进样量:5 μL)[/align][align=center][img=,558,135]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546125551_7090_2222981_3.png!w558x135.jpg[/img][/align][align=center][/align][align=left][img=,577,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101547424196_8227_2222981_3.png!w577x319.jpg[/img][/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 250 mm和高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 250 mm,以磷酸盐缓冲液(pH 6.0)-乙腈为流动相体系,在30°C柱温条件下进行梯度分析,均能够实现7-ADCA、△-2-7-ADCA、苯乙酸和扩环酸的良好分离,其中,CAPCELL PAK C[sub]18[/sub] MGII色谱柱所得峰形更佳。[/align]
做的五氯苯酚,酯化后上机2ppm 浓度峰强度仅有70000多,远低于基线600000,这是哪里出的问题,方法检出限可是0.05ppm,这是用SIM做的原因吗?
色谱柱DB23和DB-5都试过,都没有标准品峰进样口温度230,检测器温度250,柱温:60℃,以10℃/min程序升温至120℃,恒温1min,以10℃/min程序升温至200℃,恒温2min,进样量1uL;标准品为1uL苯乙醇溶于2ml乙酸乙酯溶剂峰正常,相同条件下苯乙酮标准品也有峰。为什么1-苯乙醇标准品不出峰???
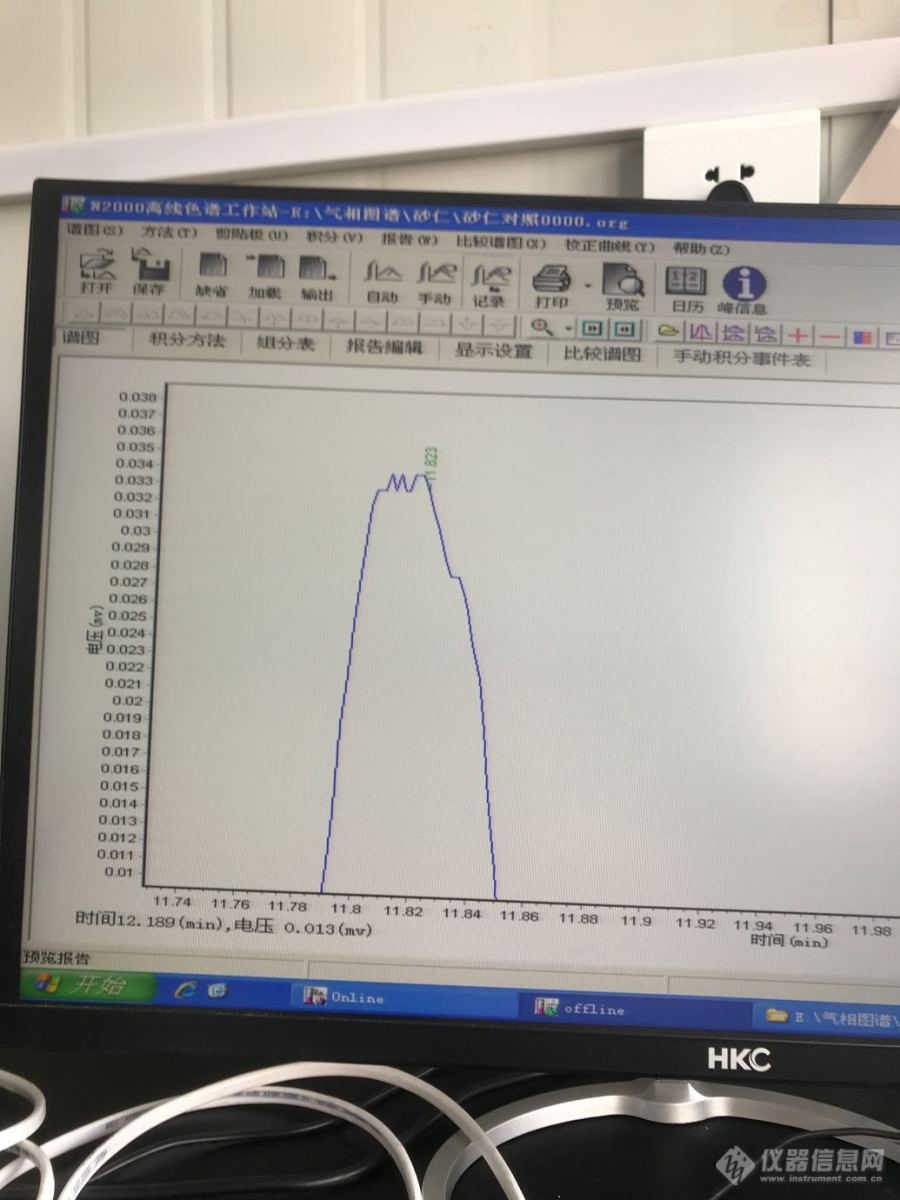
砂仁含量测定,乙酸龙脑脂对照品的峰是这样的,什么原因?[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048233492_354_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048362627_1375_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048546892_2814_4008962_3.png[/img]
国内最大最专业的国家标准物质服务平台坛墨质检-国家标准物质中心(北京坛墨质检科技有限公司),是国家质检总局指定的国家标准物质研制单位,是国内最大最专业的食品、环境、职业卫生标准物质生产商和服务商。 产品编号 产品名称 标准值 BW901263-1000-H乙腈中(易制爆)Cellulose Nitrate标准品,有证书 1000ug/mL BW901262-1000-H乙腈中氰胺/氨腈标准品,有证书 1000ug/mL BW901260-100-A-5PAK甲醇中2,4,6-三氯苯甲醚标准品,有证书 100ug/mL BW901260-100-A甲醇中2,4,6-三氯苯甲醚标准品,有证书 100ug/mL BW901259-5000-U叔丁基甲醚中2,4-二氯苯乙酸标准品,有证书 5000ug/mL BW901259-5000-A-10PAK甲醇中2,4-二氯苯乙酸标准品,有证书 5000ug/mL BW901259-200-A甲醇中2,4-二氯苯乙酸标准品,有证书 200ug/mL BW901259-2000-D-5丙酮中2,4-二氯苯乙酸标准品,有证书 2000ug/mL BW901259-2000-D丙酮中2,4-二氯苯乙酸标准品,有证书 2000ug/mL BW901259-100-U叔丁基甲醚中2,4-二氯苯乙酸标准品,有证书 100ug/mL BW901259-100-D-5PAK丙酮中2,4-二氯苯乙酸标准品,有证书 100ug/mL BW901259-100-D丙酮中2,4-二氯苯乙酸标准品,有证书 100ug/mL BW901259-100-A甲醇中2,4-二氯苯乙酸标准品,有证书 100ug/mL BW901259-1000-U叔丁基甲醚中2,4-二氯苯乙酸标准品,有证书 1000ug/mL BW901259-1000-D丙酮中2,4-二氯苯乙酸标准品,有证书 1000ug/mL BW901259-1000-A甲醇中2,4-二氯苯乙酸标准品,有证书 1000ug/L BW901258-100-A甲醇中脱乙基莠去津标准品,有证书 100ug/mL BW901258-1000-A甲醇中脱乙基莠去津标准品,有证书 1000ug/mL BW901257-5000-CL乙醇/水(1:1)中2-戊醇标准品,有证书 5000ug/mL BW901256-5000-D丙酮中邻苯二甲酸标准品,有证书 5000ug/mL BW901256-200-M二氯甲烷中邻苯二甲酸标准品,有证书 200ug/mL BW901256-100-MD二氯甲烷/丙酮(1:1)中邻苯二甲酸标准品,有证书 100ug/mL BW901256-1000-M-1二氯甲烷中邻苯二甲酸标准品,有证书 1000ug/mL BW901256-1000-M二氯甲烷中邻苯二甲酸标准品,有证书 1000ug/mL BW901256-1000-A甲醇中邻苯二甲酸标准品,有证书 1000ug/mL 坛墨质检现有员工79人,办公室面积450平米,实验室1650平米;销售、客服、财务及行政人员35人,实验室工作人员21人,库房14人,市场部8人。实验仪器设备:气相色谱、液相色谱、气质联用、液质联用、离子色谱、紫外分光光度计,原子吸收、ICP-OES和ICP-MS;库房面积450平米,库房工作人员12人,现货产品5万个,坛墨质检自主研发的产品近3000个,已申报国标345项,填补国内空白的产品达到65项。[col
用YC/T207-2014方法,测混合标液,苯乙烯 2-乙氧基乙基乙酸酯 邻-二甲苯分离不好还,出现了重合峰情况,用谱库搜出来是间二甲苯,邻二甲苯甲苯去哪里了呢?[img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221658210017_6074_5898744_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221658214942_5520_5898744_3.png[/img]
用YC/T207-2014方法,测混合标液,苯乙烯 2-乙氧基乙基乙酸酯 邻-二甲苯分离不好还,出现了重合峰情况,用谱库搜出来是间二甲苯,邻二甲苯甲苯去哪里了呢?![img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221603511087_6024_5898744_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221603511511_1108_5898744_3.png[/img]
今天做乙酸乙酯残留,对照面积分别是702.2,722.5,738.6,790.4,821.2,rsd为6.5%,第一针和最后一针面积相差120左右,但rsd在10%内,这5个对照能采用吗?
有没有老师做过苯乙醇和乙酸苯乙醇的TLG分离的。在展开剂方面给点意见吧。
一般用聚苯乙烯薄膜来校正仪器的波数,但也看到有将聚苯乙烯薄膜特定波数的吸光度归一化后作为定量计算的依据。
本人下午 用亚硝酸纳滴定,用永停滴定法 但是重氮反应较慢。不能明显的判断终点。有什么方法能明显的判断终点吗? 还有就是 中和法,用NAOH滴定能用吗? 滴定终点和化学终点偏差大吗?有哪位知道的 解答下
大家好,我们现在需要测定食品(食品包装材料)中甲苯和乙酸乙酯的测定,请大家提供一下检测方法,谢谢!
广东从日本产食品中检出甲苯和乙酸乙酯2008年10月31日 19:14来源:中国新闻网 作者:郭军 选稿:包晶晶 东方网10月31日消息:广东出入境检验检疫局今日透露,该局近日在从日本进口的日式酱油、芥末酱中检测出了甲苯和乙酸乙酯。有关食品产自三家日本生产企业。其中甲苯的最高检出值为零点零零五三mg/kg,乙酸乙酯最高检出值为零点五三七mg/kg。 此前,有日本媒体报道,日本有人食用了检出甲苯和乙酸乙酯的食品出现过不适症状。 目前,进口上述产品的中国进口企业已开始对这三家企业生产的同类产品采取下架和批批检验措施,以确保消费者安全。
先看新闻[I]新华网北京10月30日电(记者 徐博、刘铮)国家质检总局30日发布消息说,从广东出入境检验检疫机构获悉,在从日本进口的日式酱油、芥末酱中检测出了甲苯和乙酸乙酯。 国家质检总局发布的消息说,有关食品产自3家日本生产企业。其中[B]甲苯的最高检出值为0.0053mg/kg,乙酸乙酯最高检出值为0.537mg/kg[/B]。进口上述产品的中国进口企业已开始对这三家企业生产的同类产品采取下架和批批检验措施,以确保消费者安全。 此前,有日本媒体报道,日本有人食用了检出甲苯和乙酸乙酯的食品出现过不适症状。[/I]甲苯的最高检出值为0.0053 mg/kg,乙酸乙酯最高检出值为0.537 mg/kg。乙酸乙酯用途很广。主要用作溶剂,及用于染料和一些医药中间体的合成。是食用香精中用量较大的合成香料之一,大量用于调配香蕉、梨、桃、菠萝、葡萄等香型食用香精.清香型白酒是以乙酸乙酯为主体香的一类酒。其工艺特点为:清蒸、清渣、地缸发酵、清蒸馏酒。代表酒有山西汾阳杏花村。优质酒的乙酸乙酯含量为100mg/100mL以上,汾酒高达300mg/100mL,一般白酒仅含50mg/100mL,液态白酒只含30mg/100mL左右。(注意单位哦,这里是mg/100mL,换算成mg/kg大约还要乘10的)估计搞食品的,听到乙酸乙酯就笑了.哈哈!!![B]再看甲苯,[/B]《生活饮用水卫生标准》 GB 5749--20062007-07-01 实施[B]甲苯(mg/L) 0.7[/B]苯(mg/L) 0.01苯乙烯(mg/L) 0.02氯苯(mg/L) 0.3甲苯0.0053个PPM,自来水0.7PPM,这就意味着,我国的自来水的国家标准对甲苯含量的要求,比日本这个酱油里的含量要高出132倍。四个字来形容这则新闻——自取其辱。天朝进来因三聚氰胺事件恼羞成怒企图绝地反击,可惜低能到如此程度,真是让倭寇笑得合不拢嘴。
[color=#444444]我用辛酸甲酯methyloctanoate (C9H18O2) 做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的内标物,绘制不同分析物的标准曲线,各个分析物的相对校正因子差别很大。实验室人告诉我,如果分析物和内标结构差不多,那么校正因子越接近1。这是对的,不过有的化合物化学式差不多,结构却相差很多,这样校正因子差别也很大,我要如何判断我做出来的标准曲线和相对校正因子是对的呢?[/color][color=#444444]比如,我用辛酸甲酯做内标,测了两个化合物,苯乙酸(C8H8O2,含苯环和羧酸)和香兰素(C8H8O3,含苯环,羰基,甲氧基和羟基)。其中苯乙酸相对辛酸甲酯差别不是很大,而香兰素差别就大了。所以他们的标准曲线分别是y=0.7311x-0.0525 R2=0.99998,y-1.1526x-0.1764 R2=0.9982。不知道它们的校正因子对不对?有大神帮忙分析一下吗?[/color]
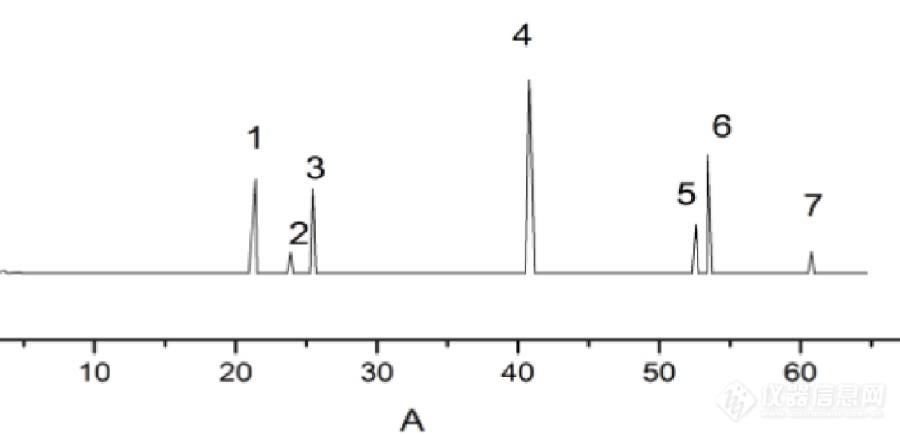
[align=center]一点红抗炎活性部位HPLC指纹图谱的研究[/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]食品事业部:石珊[/align]1 仪器与药品 岛津LC-20AT高效液相色谱仪(SPD-20A紫外检测器、Inertsil ODS-SP C18柱);BILON-1000Y超声波细胞粉碎机,上海比朗仪器制造有限公司;Infinite F50酶标仪,瑞士Tecan公司; 乙腈,色谱纯;甲酸,分析纯;槲皮素(K1505114)、槲皮苷(A1526061)、木犀草素(C10016987)、咖啡酸(I1418166)、3,4-二羟基苯乙酸(A1513031)、对羟基苯乙酸(J1409049)、对羟基苯甲酸(B1505009)、HRP(K1506040)购于阿拉丁试剂(以上对照品纯度均≥98%,可作为分析标准品);鼠李素、AA(均购自Quality Control Chemicals INC,批号分别为:10-0106/0、U-71A-S28-Z);5-LOX(recomb ined human,购于cayman公司,Batch:0467742-1);TMB-HCL西宝生物科技股份有限公司;一点红药材购自广西各大药房,样品信息具体见表1。 表1 5批一点红药材信息[table][tr][td]NO.[/td][td]生产厂家[/td][td]产地[/td][td]生产批号[/td][/tr][tr][td]1[/td][td]桂林兴安县鑫鑫中药饮片有限公司[/td][td]广西桂林[/td][td]141125[/td][/tr][tr][td]2[/td][td]广西贵港市绿之源种养发展有限公司中药饮片厂[/td][td]广西贵港[/td][td]160601[/td][/tr][tr][td]3[/td][td]广西柳州百草堂药业有限公司中药饮片厂[/td][td]广西柳州[/td][td]20151214[/td][/tr][tr][td]4[/td][td]广西柳州百草堂药业有限公司中药饮片厂[/td][td]广西柳州[/td][td]20160301[/td][/tr][tr][td]5[/td][td]广西柳州中药材市场[/td][td]广西柳州[/td][td]/[/td][/tr][/table]2 实验与方法2.1一点红提取物的制备 称取一点红粉末(过40目筛)10g,加100mL石油醚75℃ 提取2h,每小时收集滤渣。再加入100mL 70%乙醇溶液用超声波细胞粉碎机超声30min,过滤,滤渣加100 mL 70%乙醇溶解并回流4h,每2小时收集滤液,合并两次滤液,减压浓缩至无乙醇味,加水,依次用乙酸乙酯、正丁醇反复萃取,旋蒸、干燥,备用。2.2 一点红提取物抗5-LOX活性2.2.1实验原理 花生四烯酸(AA)在5-LOX催化作用下生成5-羟基氧化物(5-HPETE),5-HPETE在HRP作用下使TMB氧化,产生蓝色阳离子根,加入终止剂H2SO4后,蓝色的阳离子根转变为黄色的联苯醌,在450nm波长处检测吸光度。2.2.2 实验方法Tris-HCL:0.1M pH=7.0;5-LOX:用前取10μL 原液(6.96mg/mL),缓冲液稀释,每孔用量10μL ,终浓度为1μg/孔;AA:(0.25mg/mL)配制成21mmol/L于-20℃下保存,临用时稀释,使其终浓度为100μmol/L;HRP:配制13.9mg/mL的储备液,于4℃冰箱保存,临时用稀释20倍。TMB:配制成7.4mg/mL储备液,避光保存,临用时稀释10倍;样品制备:将提取物溶于DMSO中,配制成含1mg/mL生药储备液。 ①空白组:180μL Tris-HCL、10μL HRP ②对照组:170μL Tris-HCL、10μL 5-LOX、10μL HRP ③样品空白组:170μL Tris-HCL、10μL 药品、10μL HRP ④样品组:160μL Tris-HCL、10μL 5-LOX、10μL 药品、10μL HRP 小心震荡酶标板使反应体系混匀,37℃避光孵育10min,依次加入10μL TMB、10μL AA以激活反应,15min,加入20μL 1M H2SO4终止反应,使用酶标仪450nm处测吸光度。抑制率(%)=(△AE-△A)/△AE×100%△AE-样品池中酶与底物反应后测定的吸光度(扣除相应背景空白)△A -加入样品抑制剂后所测定的酶与底物反应的吸光度值(扣除相应背景空白)2.3 一点红抗炎活性部位指纹图谱色谱条件:流动相为1% 甲酸乙腈- 1% 甲酸水,梯度洗脱(梯度洗脱条件见表2),流速1mL/min,检测波长326nm,进样量20μL,记录色谱图65min。 表2 梯度洗脱条件[table][tr][td=3,1]t/min 1% 甲酸乙腈/% 1% 甲酸水/%[/td][/tr][tr][td]0[/td][td] 5[/td][td] 95[/td][/tr][tr][td]7[/td][td] 8[/td][td] 92[/td][/tr][tr][td]17[/td][td] 15[/td][td] 85[/td][/tr][tr][td]45[/td][td] 30[/td][td] 70[/td][/tr][tr][td]55[/td][td] 50[/td][td] 50 [/td][/tr][tr][td]60[/td][td] 70[/td][td] 30[/td][/tr][tr][td]65[/td][td] 100[/td][td] 0[/td][/tr][/table]2.3.1 对照品溶液制备 准确称取各对照品适量溶于DMSO中,按照上述色谱条件依次测定,见图1. 按出峰时间依次为对羟基苯乙酸、咖啡酸、对羟基苯甲酸、槲皮苷、木犀草素、槲皮素、鼠李素。2.3.2 供试品溶液制备 准确称取一点红乙酸乙酯、正丁醇部位10mg,DMSO溶解,配制成1mg/mL溶液。按上述色谱条件测定。2.3.3 精密度试验 取同一供试品溶液连续进样6次,考察被确认共有峰的保留时间和峰面积。结果表明,各色谱峰的保留时间的RSD1%,峰面积的RSD3%。2.3.4 稳定性试验 取同一供试品溶液,分别在0,3,6,9,12,24h进样检测,考察被确认共有峰的保留时间和峰面积。结果表明,各色谱峰的保留时间RSD1%,峰面积RSD3%。2.3.5重复性试验 取同一供试品溶液5份,按上述方法制备和检测,考察被确认共有峰的保留时间和峰面积。结果表明,各色谱峰的保留时间RSD1%,峰面积RSD3%。2.3.6供试品溶液的制备 准确称取5个批次的一点红粉末(过40目筛)5g,加50%甲醇100 mL超声(40HZ,200W)提取60min,过滤收集滤液定容至100 mL,用0.45μm微孔膜滤过,待用。3结果3.1 一点红提取部位抗5-LOX活性 不同产地的一点红乙酸乙酯、正丁醇部位对5-LOX均有抑制作用。结果表明,一点红药材正丁醇提取部位抗5-LOX活性略高于乙酸乙酯部位。 表3为一点红不同活性部位抗5-LOX 活性[table][tr][td][align=center]厂家[/align][/td][td] 乙酸乙酯部位 正丁醇部位[/td][/tr][tr][td=2,1] 洪才 60±0.1% 64±0.1%[/td][/tr][tr][td=2,1] 绿之源 59.2±0.1% 65.6±0.1%[/td][/tr][tr][td=2,1] 百草堂(1) 59.4±0.1% 65±0.1%[/td][/tr][tr][td=2,1] 百草堂(2) 59±0.1% 65.2±0.1%[/td][/tr][tr][td=2,1] 中药材市场 59.5±0.1% 64.6±0.1%[/td][/tr][/table]3.2一点红乙酸乙酯部位HPLC指纹图谱与相似度分析 分析一点红乙酸乙酯部位和正丁醇部位HPLC指纹图谱,发现乙酸乙酯部位图谱重现性、精密度、稳定性良好,且该部位化学信息较多。确定了该部位7个色谱峰,按保留时间先后顺序依次为:对羟基苯乙酸(21.7min)、咖啡酸(25.1min)、对羟基苯甲酸(25.6min)、槲皮苷(41.0min)、木犀草素(52.7min)、槲皮素(53.3min)、鼠李素(60.8min)。其中,4号槲皮苷含量最多,6号槲皮素次之,1号对羟基苯乙酸含量最少(见图1)。[align=center][img=,690,333]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091742404389_6508_2904018_3.png!w690x333.jpg[/img][/align][align=center][img=,593,315]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091742523769_7471_2904018_3.png!w593x315.jpg[/img] B[/align]A 对照品HPLC图 B 乙酸乙酯部位HPLC图1.对羟基苯乙酸 2.咖啡酸 3.对羟基苯甲酸 4.槲皮苷 5.木犀草素 6.槲皮素 7.鼠李素 图1 一点红对照品混合溶液色谱图及其指纹图谱共有模式3.2.2 5批一点红药材乙酸乙酯部位样品溶液测定 对5个批次的一点红药材按上述方法提取得到乙酸乙酯部位并进行HPLC分析,得到5个批次一点红乙酸乙酯部位指纹图谱(见图2)。利用“中药色谱指纹图谱相似度评价系统2004A版”进行相似度评价,共标定了18个特征峰作为判别一点红抗炎部位质量的群体特征峰。不同来源一点红的HPLC乙酸乙酯部位指纹图谱相似度评价见表4。[align=center][img=,690,335]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091743032379_5760_2904018_3.png!w690x335.jpg[/img][/align] 图2 5批一点红乙酸乙酯部位HPLC指纹图谱 表4 不同厂家一点红药材相似度评价结果[table][tr][td] [/td][td][align=center]1[/align][/td][td][align=center]2[/align][/td][td][align=center]3[/align][/td][td][align=center]4[/align][/td][td][align=center]5[/align][/td][/tr][tr][td]厂家[/td][td][align=center]洪才[/align][/td][td][align=center]绿之源[/align][/td][td][align=center]百草堂(1)[/align][/td][td][align=center]百草堂(2)[/align][/td][td][align=center]中药材市场[/align][/td][/tr][tr][td]相似度[/td][td] 0.971[/td][td] 0.982[/td][td] 0.988[/td][td] 0.973[/td][td] 0.895[/td][/tr][/table]4.讨论早期研究表明,一点红水提物和甲醇提取物与阿司匹林相比,对蛋清蛋白引起的小鼠足肿胀有较好的抑制作用[sup][/sup]。一点红甲醇提取物及二氯甲烷提取物可通过抑制炎性介质和细胞因子IL-1β、TNF-α细胞因子和NO来减轻炎症反应[sup][/sup]。一点红在临床上用于治疗静脉炎效果强于喜疗妥软膏,是治疗静脉炎的理想药物[sup][/sup]。本实验通过一点红乙酸乙酯部位对5-LOX的抑制,从而阻止5-LOX途径,减轻炎性症状。本实验对比了超声波细胞粉碎0min、60min、120min乙酸乙酯、正丁醇提取率,表明随着超声时间的延长,石油醚、乙酸乙酯部位提取率逐渐减小,正丁醇部位几乎无影响。可能由于细胞粉碎时间过长,导致糖类、粘[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]等杂质的浸出,并附着于药材表面,影响乙醇的进入,从而影响石油醚、乙酸乙酯部位的提取率。考察乙醇加热回流2h、3h、4h,1h收集滤液以及回流4h,2h收集滤液。发现回流4h,2h收集滤液,各部位提取率最高。5-LOX体外抑制实验表明,一点红乙酸乙酯部位和正丁醇部位均具有良好的抗5-LOX活性,由于乙酸乙酯部位所含化学信息多,且谱图重现性、精密度、稳定性均良好。故建立了一点红乙酸乙酯部位HPLC指纹图谱,标定了18个共有峰,确定了该部位7个色谱峰,按保留时间先后顺序依次为:对羟基苯乙酸、咖啡酸、对羟基苯甲酸、槲皮苷、木犀草素、槲皮素、鼠李素。
我现在在做某物质的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]残留溶剂,标准规定乙酸丁酯≤1000ppm,我配置对照品溶液时可以配置的比标准规定的低吗(比如对照品溶液配成500ppm)?样品中乙酸丁酯的残留大概是80ppm
======在 2006-11-13 13:46:31 wangjiangli0123来信中写道:======请问 电离电位是多少 苯乙烯 的 乙酸丁酯的 还有十一烷的======================================
请教 氯乙酸 苯酚 苯氧乙酸用什么做展开剂?即氯乙酸和苯酚反应生成苯氧乙酸。
苯乙酸的气相检测条件--请高手帮忙!哪位大侠知道苯乙酸气相检测,用什么柱子,条件是什么?谢谢啦!
问题:现在想测试植物叶(研磨成粉末)中有机酸,如乙酸、丙酸、异丁酸、丁酸、异戊酸、壬酸、苯甲酸、月桂酸、苯乙酸、草酸、柠檬酸、苹果酸等其他有机酸。想用GC-MS 测试,现在有HP-FFAP(30*0.25*0.25)柱子。样品处理:想用一定的酸度的强无机酸溶液萃取样品,然后用CH2Cl2进行萃取,然后上机测试,不知道可否?如果CH2Cl2不行,请问用什么溶剂进行萃取好?或者怎样对样品进行前处理(指不需要酯化)。如果一定要酯化,有简单的酯化方法不?测试方法:如果用该柱子,测试参数如何设定呢?实在迷惑,有着急完成任务,望专家指导,谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP