求哪位大虾帮忙下2个工业级化学品的质量标准,品种为:异丁醛、丁酮。在国标网有找到,不过要花mm,哪位大虾有现成的,帮帮忙了,先在此谢过。
唐访良 朱文(杭州市环境监测中心站 杭州 310007) 摘 要:用活性炭吸附环境空气和废气中的2-丁酮和苯,经二硫化碳解吸后用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定。方法的回收率:2-丁酮为86.2%~104.6%,苯为91.3%~105.7%;变异系数:2-丁酮为2.5%~2.8%,苯为2.5%~3.8%。当采样体积为20L,解吸液体积为2.00mL,进样体积为2μL时,2-丁酮和苯的最低检测浓度分别为0.03mg/m3和0.01mg/m3。 关键词:环境空气和废气 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url] 2-丁酮 苯 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url] DETERMINATION OF 2-BUTANONE AND BENZENE IN AMBIENT AIR AND EXHAUST AIRTANG Fang-liang and ZHU Wen(Hangzhou Environmental Monitoring Centre, Hangzhou 310007,China) Abstract:Sampling of micro-amounta of 2-butanone and benzene in ambient air and exhaust air was carried out by their adsorption on active carbon filled in a sampling tube, and then desorbed with 2.00ml of carbon disulfide. Two μL of the desorbed solution were taken and analyzed by gas chromatography. Satisfactory separation was attained by using PEG-20M column. Retention time method was used in the qualitative analysis and peak height method was used in the quantitative analysis. Mixed standard solutions containing 2-butanone from 0.00 to 64.8mg• L-1 and benzene from 0.00 to 70.4 mg• L-1(in CS2) were used to prepare an external standard curve. RSD’s(n=8) for 2-butanone and for benzene were found to be in the range of 2.5%~2.8% and 2.5%~3.8% respectively. Recoveries found for 2-butanone and for benzene were in the range of 86.2% to 104.6% and 91.3% to 105.7% respectively. The detection limits(S/N=2) were found as 0.03mg• m-3(for 2-butanone and 0.01mg• m-3(for benzene) under the experimental conditions mentioned above and when 20L of sampling volume were taken. Keywords:Gas chromatography 2-Butanone Benzene Ambient air Exhaust air 2-丁酮和苯都是常用的有机溶剂,应用广泛。由于苯有毒有害,是人类公认的致癌物之一,在生产和使用过程中不可避免地会造成对人体的损害和对环境的污染[1]。随着人们环保意识的提高,人们已越来越多的用低毒或无毒溶剂来代替毒性大的溶剂,2-丁酮是常用的替代品之一,如有些胶粘剂所用的溶剂,就有用2-丁酮来代替苯、甲苯、二甲苯作为溶剂的。环境空气和废气中的苯常用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定[2],车间空气中2-丁酮也有用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法进行测定的报道[3],但环境空气和废气中2-丁酮和苯的同时分离和测定还较少报道。本文用活性炭吸附富集环境空气和废气中2-丁酮和苯,经二硫化碳(CS2)解吸后[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定,方法简便、快速,用于实际样品测定,结果满意。1 试验部分1.1主要仪器和试剂 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-9A[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](日本岛津公司),具FID检测器,KB-6A型大气采样器(青岛市崂山电子仪器实验所),2L铝箔复合膜软气袋(简称气袋),活性炭吸附采样管(市售,内填0.2g粒度为20~40目的粒状活性炭);苯、甲苯、对-二甲苯、间-二甲苯、邻-二甲苯皆为色谱纯试剂,2-丁酮、丙酮为分析纯试剂;二硫化碳(CS2)为分析纯试剂,经纯化处理后用作溶剂。1.2色谱条件 色谱柱为长2m内径3mm的玻璃柱,内填10%PEG-20M/Shimalite W (NAW)(60~80目);柱温为70℃,汽化室、检测室温度为150℃;载气(N2)流速为50mL/min ,燃气(H2)流速为45 mL/min ,助燃气(A ir)流速为500 mL/min 。进样量为2μL1.3样品的采集和测定 环境空气样品中的2-丁酮和苯的浓度一般较低,常需用活性炭吸附采样管进行富集浓缩,按文献[2]进行环境空气样品的采集,按文献[4]进行污染源废气样品的采集。将上述已采样的活性炭采样管中的活性炭移入10mL具塞比色管中,加2.00mLCS2,轻轻振摇2min,放置20min后,吸取2μL解吸液,进行分离测定。以保留时间定性,以峰高外标法定量。 2 结果与讨论2.1色谱柱的选择 2-丁酮的沸点为79.6℃,苯的沸点为80.1℃,二者沸点相差0.5℃,用文献[2]给出的2.5%DNP+2.5%Bentone-34和PEG-400柱分析2-丁酮和苯,二者不能分离,本文选用PEG-20M柱,2-丁酮和苯能很好分离,且分析样品所需的时间短,如图1所示。2.2干扰试验 选择与2-丁酮、苯经常同时使用或共存的丙酮、甲苯、对-二甲苯、间-二甲苯、邻-二甲苯进行干扰试验(如图1)。从图中可以看出,在选定的色谱条件下,各组分之间能很好分离,不会干扰2-丁酮和苯的分离和定性、定量测定。 图1 2-丁酮和苯与共存物质的分离色谱图 图中 1.溶剂(CS2,0.59分) 2.丙酮(0.74分) 3.2-丁酮(1.19分) 4.苯(1 .56分) 5.甲苯(2.79分) 6.对-二甲苯+间-二甲苯(5.14分) 7.邻-二甲苯(6.74分)2.3 标准曲线及精密度检验 配制2-丁酮浓度为0.00,6.48,13.0,25.9,38.9,51.8,64.8mg/L和苯浓度为0.00,7.04,14.1,28.2,42.2,56.3,70.4mg/L的CS2混合标准系列,在选定的色谱条件下进行测定,线性回归方程和相关系数:2-丁酮,H=31.3C-15.1(C为mg/L ),γ=0.9999;苯,H=45.9C-4.35(C为mg/L),γ=0.9999。以混合标准系列浓度最高点的0.1倍(即2-丁酮浓度为6.48 mg/L、苯浓度为7.04 mg/L)和0.9倍(即2-丁酮浓度为58.3 mg/L、苯浓度为63.4 mg/L)的标准溶液为样品,各重复8次试验, 其变异系数: 0.1倍溶液,2-丁酮和苯分别为2.8%、3.8%; 0.9倍溶液,2-丁酮和苯皆为2.5%。2.4采样效率试验 采用相对比较法来评价采样效率。在2L气袋中用高纯氮气配制了不同浓度的2-丁酮和苯的混合气体样品,串联2支活性炭采样管,第一管连气袋,第二管连大气采样器,如1.2进行样品的采集和测定,结果如表1。表1 采样效率试验测定结果 序号 项 目 第一管测定量(m/μg) 第二管测定量(m/μg) 第一管采样效率( %) 1 2-丁酮 8.18 0.00 100 苯 5.27 0.00 100 2 2-丁酮 70.3 0.00 100 苯 49.0 0.00 100 3 2-丁酮 900 0.00 100 苯 177 0.00 100 结果表明,在上述测定量条件下,第一支采样管的采样效率已达100%,用一支采样管已能满足测定要求。 2.5 方法的检出限按两倍噪声计算的仪器的最小检出量,2-丁酮为0.6ng,苯为0.2ng (进样体积为2μL),当采样体积为20L,解吸液体积为2.00mL时,方法的最低检出浓度:2-丁酮为0.03mg/m3,苯为0.01mg/m3。2.6 样品的测定和回收率用本法测定了环境空气样品和某污染源废气样品,并在解吸液中进行了加标回收试验,结果如表2。 表2 样品测定结果和回收率 样品名称 项 目 样品含量(m/μg) 加标量(m/μg) 测得总量(m/μg) 回收率 (%) 环境空气 2-丁酮 0.00 0.65 0.68 104.6 苯 0.32 0.70 1.06 105.7 废气 2-丁酮 1620 1396 2824 86.2 苯 7.04 6.42 12.9 91.3
求助标3-邻苯二甲酰亚胺-丁酮的准红外光谱,CAS号为2028-33-3,英文名 N-(1-methyl-2-oxo-propyl)-phthalimide,
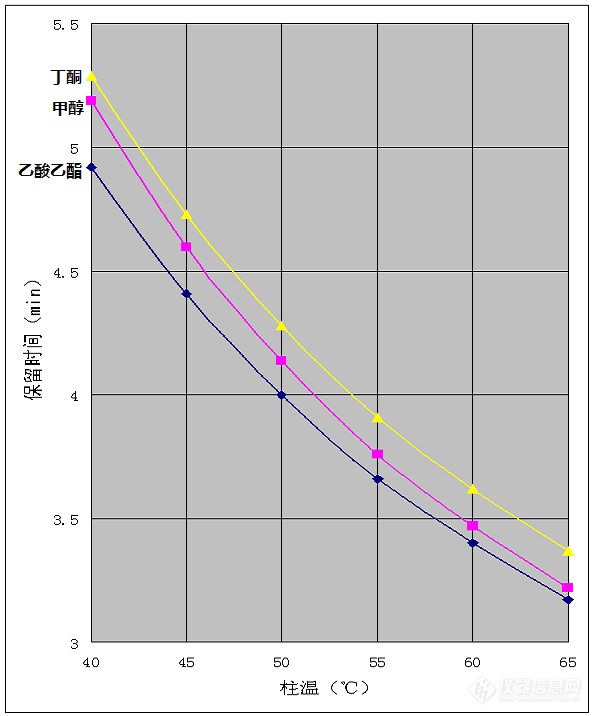
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。
如题:如何检测微量丁酮和甲苯的含量请楼主说明是什么样品?
如何测试二甲苯,丁酮,甲苯,乙苯醇这四个的浓度?
请教高手:有可以同时测2-甲基甲醛、甲苯、丁酮的毛细管柱吗?用GCMS检测。谢谢!
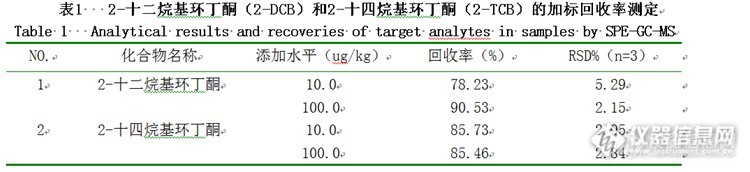
摘 要:建立固相萃取-气相色谱- 质谱联用(solid phase extraction with gas chromatography-mass spectrometry,SPE-GC-MS)法测定植物油中2-十二烷基环丁酮和2-十四烷基环丁酮。对影响分析物萃取效率的诸因素如洗脱溶剂等进行详细考察和优化。最佳萃取条件为0.5 g样品与5 mL乙腈混匀,经ProElut Silica (500 mg/3mL)固相萃取柱净化后,以GC-MS 进行测定,该方法对2-十二烷基环丁酮和2-十四烷基环丁酮的检出限为10μg/kg,线性范围为0.01~0.5μg/mL,线性相关系数分别为0.99938和0.99977,相对标准偏差(relative standard deviation,RSD)(n=3)小于6%。该方法成功应用于植物油中2-十二烷基环丁酮和2-十四烷基环丁酮的分析,加标回收的回收率为78%~91%。关键词:固相萃取;气相色谱-质谱;2-十二烷基环丁酮;2-十四烷基环丁酮;植物油 食品辐照作为对物质或食品进行加工处理的新型保藏技术,在国际上已逐渐被认可,但是在商业化应用、国际贸易以及辐照食品的市场监管方面,迫切需要有辐照食品鉴定检测方法。 经辐照后,在含脂食品中会产生特异性辐解产物2-烷基环丁酮(2-Alkylcyclobutanones ,2-ACBs),它是含脂辐照食品的特异性辐解产物,在未辐照的含脂食品中,至今还从未检测到此类化合物。在1990年, 2-ACBs 类化合物可作为检测含脂辐照食品的标志性化合物, 首次被报道,随后依据该结论制定了欧盟标准EN1785和GB\T 21926-2008 。2-ACBs由食品中的游离脂肪酸或甘油三酸酯的羰基氧失去一个电子,再经由重排过程生成,其过程如图1所示。http://ng1.17img.cn/bbsfiles/images/2015/07/201507091523_554630_2452211_3.png图1 经辐照后游离的脂肪酸转化为2-ACBs的示意图 在大多数食品中,棕榈酸、硬脂酸、油酸、亚油酸是主要的脂肪酸,而棕榈酸和硬脂酸是其中含量最高的饱和脂肪酸,其辐解物2-十二烷基环丁酮(2-dodecylcyclobutanone,2-DCB)和2-十四烷基环丁酮(2-tetradecylcyclobutanone,2-TCB)相对于其它2-ACBs较为稳定,因此一般作为检测含脂辐照食品的主要标志性化合物。目前对含脂辐照食品大多采用佛罗里硅土柱进行净化,但是该法的应用范围有限。本实验拟通过优化固相萃取(solidphase extraction,SPE)条件,采用气相色谱-质谱联用(gas chromatography-massspectrometry,GC-MS)技术测定植物油中2-十二烷基环丁酮和2-十四烷基环丁酮,为进一步缩短2-ACBs 萃取和分离时间、减少溶剂使用量、提高检测灵敏度以及扩大方法应用范围提供基础数据和理论依据。1 材料与方法1.1 材料、试剂与仪器GCMS-QP2010 气相色谱-质谱联用仪 日本岛津公司;DM-5MS 毛细管柱(30 m×0.25 mm,0.25 μm)迪马公司;XH-C 涡旋混合器 江苏金坛市盛威实验仪器;80-1 高速离心机 河南省予华仪器;OSB-2100 旋转蒸发仪 上海爱朗仪器有限公司;12孔固相萃取装置 迪马公司; ProElut Silica(500 mg/3mL)固相萃取柱 迪马公司。HSC-12B 氮吹仪天津市威仪科技发展有限公司;丙酮、二氯甲烷、乙酸乙酯乙腈、甲基叔丁基醚、正己烷(均为色谱纯)迪马公司。实验所用的植物油均购自当地市场。1.2 方法1.2.1 标准贮备液的制备称取一定量标准品,溶于正己烷溶剂中,配制成浓度为0.5 mg/mL的标准贮备液。再配制成质量浓度系列为0.01μg/mL、0.02μg/mL、0.05μg/mL、0.1μg/mL、0.2μg/mL、0.5μg/mL的标准工作溶液,备用。1.2.2 仪器分析条件气相色谱条件:色谱柱为DM-5MS (30.0m×250μm,0.25μm);载气He(99.995%);恒流,柱流速1.0mL/min;不分流,进样量1μL,进样口温度为260℃;起始温度80℃(保持1min),以15℃/min的速度升至150℃,再以8℃/min升温至200℃,再以20℃/min升温至260℃(保持5min)。质谱条件:EI源,离子源200℃,溶剂延迟为3min,选择离子监测模式(SIM),选择监测离子(m/z):69、84、98、112、125。1.2.3 样品的提取称取0.5 g样品于10 mL带塞试管中,加入5 mL乙腈,涡旋混合2 min,超声提取2 min,4000 rpm下离心2min,取上清液;下层油脂再用5 mL乙腈重复上述步骤,合并两次上清液。将得到的上清液在50℃下,氮吹近干,再慢慢挥干,再向氮吹瓶中加入2.5 mL正己烷复溶,待净化。1.2.4 样品的净化依次用5 mL甲基叔丁基醚,5mL正己烷缓慢通过ProElut Silica固相萃取柱,以达到润湿小柱,活化填料,除去干扰杂质的目的;再将1.2.3节方法制得的待净化液转移到ProElut Silica固相萃取柱中,流出液弃去;然后用5 mL正己烷淋洗,弃去流出液;再用10 mL甲基叔丁基醚:正己烷(1:99V:V)洗脱,用旋转蒸发瓶接收,直至洗脱液完全自然滴出。在50 ℃下,将收集到的洗脱液氮吹浓缩,然后用正己烷定容至1 mL后供GC-MS分析。2 结果与分析在固相萃取操作中,影响分析物峰面积的主要固相萃取因素有洗脱剂、洗脱体积、洗脱速率和上样速率。为了获得最佳分析结果,需要对其进行优化。2.1固相萃取条件的确定2.1.1 提取溶剂的选择2-十二烷基环丁酮(2-DCB)和2-十四烷基环丁酮(2-TCB)与脂肪酸的结构及其类似,故能溶于极性和中等极性的试剂中。分别用丙酮、二氯甲烷、甲基叔丁基醚、乙酸乙酯作为2-DCB 和2-TCB的提取溶剂。实验结果表明乙腈提取效果较好,再加以涡旋振荡后结合超声提高回收率。2.1.2 固相萃取柱的选择对于油脂类样品,采用固相萃取柱进行样品净化是必不可少的步骤。结合相应参考文献,本实验采用了硅胶、PSA、Florisil、Alumina等填料的固相萃取柱,结果表明对于植物油,硅胶柱相对于其他填料的固相萃取柱来说,2-DCB 和2-TCB回收率较高,添加回收率达到了80%-120%,满足分析检测的要求,且达到很好的净化效果。如图2所示http://ng1.17img.cn/bbsfiles/images/2015/07/201507091524_554631_2452211_3.pngA:标准品;B:空白样品;C:添加标品的样品图2 植物油空白样品及其添加样品的总离子流图2.1.3 淋洗曲线的建立固相萃取技术最重要的目的在于通过固相萃取柱将目标化合物与主要干扰物分开,从而实现净化的目的。在此过程中应非常注意选择合适的洗脱溶剂。样品处理过程是先用正己烷将其中的中性化合物除去,参照Horvarovich 等报道,用硅胶柱分离样品中的2-DCB和2-TCB,选用弱极性的甲基叔丁基醚(methyl-t-butyl ether,TBME)/正己烷(V/V)混合溶剂将稍强极性的2-DCB 和2-TCB洗脱下来。由于样品基质与文献不一样,淋洗液与洗脱液的选择也会不一样。因次需要考察正己烷以及其与甲基叔丁基醚不同比列的混合液作为洗脱液时2-DCB和2-TCB的回收率。选用5根ProElut Silica固相萃取柱,取0%、0.5%、1%、2%、5%不同浓度的甲基叔丁基醚:正己烷(V/
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.
为了达到某些需求,很多时候单一的溶剂并不能满足挥发速率等方面的要求,故产生了对混合溶剂的需求,然而现在的一些混合溶剂的气味很大,如中干水、洗网水、硝基稀料等,他们大多是甲苯、环己酮、丁酮等混合而成,如何才能将这种混合溶剂的气味降低呢,还请各位多多指教!不胜感激!
用甲醇钠制得丁酮烯醇钠,计算收率的时候用什么方法来确定干燥品中丁酮烯醇钠的占比是多少呢?
哪位有工业用丁酮的国家标准,可以提供给我一份吗?或者告诉我在哪里可以找的到?不胜感激!另外还想问一下,像中干水、硝基稀料这样的混合物有国家标准吗?
在某网上看到一段话:关于丁酮与丙酮的区别描述:丙酮是三个碳原子,丁酮是四个碳原子,都有羰基,丙酮易挥发而丁酮挥发性没有丙酮好丁酮比丙酮毒性低.丙酮作为溶剂最大的缺点就是挥发性太强,这也是它无法替代丁酮的最主要的原因.丁酮沸点 73.4 ℃丙酮沸点56.05℃大家觉得这些描述正确吗?还有什么补充的吗?最根本的区别是这个吗?
HPINNOWAX60米色谱柱分离甲醇,丁酮,怎么设置条件都分不开,安捷伦工程师说可以在35度或者更低的条件下保持一段时间,单纯靠物质沸点分离,可是尝试后依旧分不开,大家试过这种方法吗?
RT,纯的丁酮(甲乙酮)的电导率很低,大概是0.07ms/m,我想吧它的电导率提高到1.5ms/s,要怎么样才能达到?我试了加入NaCl水溶液,发现根本不行,丁酮只能溶解一点点水,结果电导率提高的很少,只到0.6ms/m,就很难再提高了。能不能加入其他什么物质,本身电导率很高,既能和丁酮互溶呢?
最近要测试丁酮的含量,各位有无做过呀?GC-MS仪器参数怎样设置?
请教: 1.在KB-WAX柱子上进丁酮标液,我将柱温分别设为120、160、180度,发现丁酮出峰时间为3.60min、3.71min、3.83min,不是应该柱温越高,出峰越早吗? 2.为什么在不同柱子上,同一种物质,出峰顺序不一样? 如:TVOC标液在苯填充柱上,苯乙烯在邻二甲苯之后出峰;而在TVOC柱子上苯乙烯却在邻二甲苯之前出峰?
[color=#333333]我用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]给乙酯与丁酮打色谱算含量,结果发现都是99.99%,这个结果肯定不对,后来,我直接各50%混合,再次打色谱,结果还是99.99%并没有预想的50%:50%,这是怎么回事啊,乙酯,丁酯单独打色谱,出峰时间很相似都是1分钟多出峰了,柱箱温度都是65,进样温度150,检测温度150,是否有错误,还请帮忙解答,谢谢您了。 [/color][color=#333333]乙酯,丁酮单独做色谱分析是,乙酯出峰时间为1.090分钟,丁酮出峰时间为1.148分钟[/color]
热解吸做voc中丁酮和甲醇峰分不开?用的是wax柱子,请问各位老师还能怎么分离开?第一个峰是丙酮,第二峰是乙酸乙酯,第三个峰是溶剂甲醇峰覆盖目标丁酮。[img]https://ng1.17img.cn/bbsfiles/images/2018/11/201811171633448269_6611_3333718_3.png[/img]
甲酮与丁酮是同一样试剂,你认为对吗?为什么?
关于丁酮肟和水的分离,有没有什么好方法?或者有什么萃取剂?用的异辛醇总有一部分水在有记层。
我用AE.SE-30检测甲基三丁酮肟基硅烷和乙烯基三丁酮肟基硅烷,这两种物质用的毛细管色谱柱检测不出来,出的都是连在一起向上漂移的峰。请教用什么样的柱子检测?
今天来了一个原料,3-巯基-2-丁酮,GC-MS检测的结果如附件。我想咨询一下,后面RT17.275的峰是怎么回事呢?是否是3-巯基-2-丁酮的聚合产物?
请教各位: 我将丙酮和丁酮混标分别注入FFAP、TVOC、,均能得到较好的分离条件,但做职业卫生样品时发现FFAP柱子在丙酮或丁酮出峰时间出的峰很大,有的时候甚至能超标,但同一个样品用TVOC柱子不会有这种情况,测得的值较低。请问这是怎么回事? 现在我又将丙酮和丁酮混标 打入KB-WAX柱子,峰形也很好,会不会再出现上述情况?与柱子极性有关系吗?
分享一篇利用在磁性管内的固相微萃取技术结合[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法测定血清中己醛和2-丁酮的技术。该技术创新点在于制备了一种新型的具有Fe3O4核和C8功能化介孔二氧化硅壳的磁性介孔吸附剂,并利用2,4-二硝基苯肼预载在聚四氟乙烯管内,在磁场的作用下用聚四氟乙烯管从人血清中萃取痕量的目标物,同时在吸附剂表面产生衍生物。大量的血清样本不需经过预处理,吸附剂可直接用于提取。
想要分析水中溶解的挥发性物质,丁酮及甲醇。我使用的机子是岛津GC-17A,inj:110,col:40度,2度/分到50度,50度保持时间5分,DET:300度。出来的峰重合在一齐,不知道有什么方法让他们分离!!
今天我用SPB-5(岛津的GC2010)去做空气中的丙酮和丁酮,无奈怎样调节各个温度也不能分得开,老是二硫化碳相近得多,要求高人指点一下。
据瑞士联邦政府网站报道,瑞士官员日前表示,当局发现圣安德里(San Andrea)牌两瓶矿泉水中含丁酮溶剂后,已下令将这类矿泉水全面下架。瑞士公共卫生局在声明中表示:"回收这商品是因在两瓶两公升矿泉水中发现丁酮溶剂。"并进一步表示,无论有无气泡,这类矿泉水一律下架。声明中指出,这项无色产品"一开瓶就会闻到溶剂难闻气味"。
有机化学丁酮和醇钠反应的主产物并说明理由
[color=#444444]请问4-羟基,2-丁酮的检测方法?,可以具体到用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的什么柱子,进样温度吗?[/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP