有老师按照中国兽药典2020年一部,丙二醇的乙二醇含量的项目吗?按照药典的方法设置,对照品溶液中乙二胺的峰没有出!不知道怎么回事?
甘油乙二醇二甘醇图为3针对照品,完全不平行,且二甘醇峰(14.5左右)越来越小……做了很多遍,都是这样,先后用db_624,0.53*3,ov_1301,0.25*1.4柱子试过,二甘醇不是和甘油包一起,就是这样不怎么出峰,而且面积也并不平行,求各位大神看看,指点一下??
如题,有朋友用的是ZB624柱(固定液:6%氰丙基苯基-94%二甲基聚硅氧烷),柱长30m,内径530um,膜厚0.3um。载气流速4.5ml/min。其他条件同10版药典。结果发现,对照品溶液图谱情况良好,各峰分离度很好,峰面积之比(该法为内标法测定)的RSD5%,出峰顺利乙二醇-正己醇-二甘醇。但系统适应性图谱就不行了,溶剂峰(甲醇)拖尾严重,干扰乙二醇,更纳闷的是二甘醇峰会消失,在二甘醇应该出现的保留时间前后也无峰出现,加大二甘醇的浓度也没有峰出现,真是怪了。做了好多次了,都是如此。可以确定的是,做供试分析会严重污染柱子,对照品溶液(无甘油)进样分析正常(多次进样也正常),供试品溶液和系统适应性样品溶液(均含甘油)分析溶剂峰就会拖尾,影响乙二醇峰,然后再进对照品溶液也会如此。但柱子老化后,对照品溶液进样分析就会正常了。请教做过这个品种的同行,有没有遇到这问题?这种情况可能是那里出现了问题?
乙二醇、二甘醇和三甘醇取本品4g,精密称定。置 100ml量瓶中,取1,3-丁二醇0.004g,精密称定,置同一量瓶中,加丙酮使溶解,相同溶剂稀释至刻度,作为供试品溶液。取乙二醇0.0025g, 二甘醇0.004g,三甘醇0.004g,精密称定,置同一100ml量瓶中,取1,3-丁二醇0.004g,置 该量瓶中,加丙酮使溶解,相同溶剂稀释至刻度,作为对照品溶液。照[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法(通则0521)试验。以50%苯基-50% 甲基聚硅氧烷为固定液(液膜厚度1.0μm)的毛细管柱,起始 温度为40℃,以每分钟10℃的速率升温至60℃,维持5分钟,再以每分钟10℃的速率升温至170℃,维持0分钟,再以每分钟15℃的速率升温至280℃。维持60分钟(可根据具体情况调整)。检测器为氢火焰离子化检测器。检测器温度 290℃,进样口温度为270℃。取对照品溶液作为系统适用性 试验溶液,载气为氦气,流速5.0ml/min,分流比2:1,进 样体积1.0μl。乙二醇,二甘醇和三甘醇与内标1,3-丁二醇的分离度均不得小于2.0,各峰间的拖尾因子应符合规定,[color=#ff0000]乙二醇,二甘醇和三甘醇峰面积相对于内标1,3-丁二醇的峰面积相对标准偏差不得过5.0%[/color]。以1,3-丁二醇峰面积计算乙二醇,二甘醇和三甘醇的峰面积,以下式计算:[color=#ff0000]结果=(Ru / Rs) X (Cs X Cu)X F X100 [/color] 式中Ru为供试品溶液中各待测物质与内标的峰面积比率;Rs为对照品溶液中各对照物质(乙二醇,二甘醇和三 甘醇)与内标的峰面积比率; Cs为对照品溶液中各对照物质(乙二醇,二甘醇和三 甘醇)的浓度,ug/ml;Cu为供试品溶液中待测物质的浓度,mg/ml F为转换因子,103mg/g。(10的三次方 )依法检测,乙二醇,二甘醇和三甘醇均不得过0.01%。以上标红文字不能理解其含义,望解答,谢谢 !

检测甘油,有对照乙二醇、二甘醇,内标正己醇,条件:DB-624(30m*0.53mm 3.um),进样量1ul,分流比10:1,进样口200°,FID250°,程序:起始100(维持4′),以50°/min升120°(维持10′),再以50°/min升220°(维持6′).后加设降温和平衡时间。样品处理:系统适用性性乙二醇、二甘醇,内标正己醇各100mg稀释至100ml(系统储备液),精取1ml+4g甘油样品至100容量瓶, 所 有 溶剂都是色谱甲醇。 对照液:乙二醇、二甘醇,内标正己醇各50mg至100ml(标储液),取5ml稀释至25ml。问题:6月份同样方法检测,一切正常(当时柱子新买来活化后检1批乙醇,) 这两天同一根柱子检测(中间检测了3批乙醇),结果系统适用性乙二醇出不来峰了。正己醇和二甘醇峰面积无论是储备液还是系统适用性都没什么差异。储备液中的乙二醇有峰面积,但与之前浓度相当情况下峰面积小1/3,系统适用性就出不来了,对照液要算校正因子f,之前差不多2-3左右,现在超过10了。乙二醇的安剖瓶色标5ml有之前开启后密封冷藏的,也有新开的,两种情况都差不多,批号都是081226。http://ng1.17img.cn/bbsfiles/images/2012/07/201207251613_379663_2481522_3.jpg6月份的对照液,峰依次是:乙二醇--正己醇--二甘醇。http://ng1.17img.cn/bbsfiles/images/2012/07/201207251616_379664_2481522_3.jpg6月的系统适用性,7.8′乙二醇峰还是不错的,但这次此峰消失了。后面的正己醇、二甘醇相当浓度峰面积也相当。请问问题可能在哪里呢?
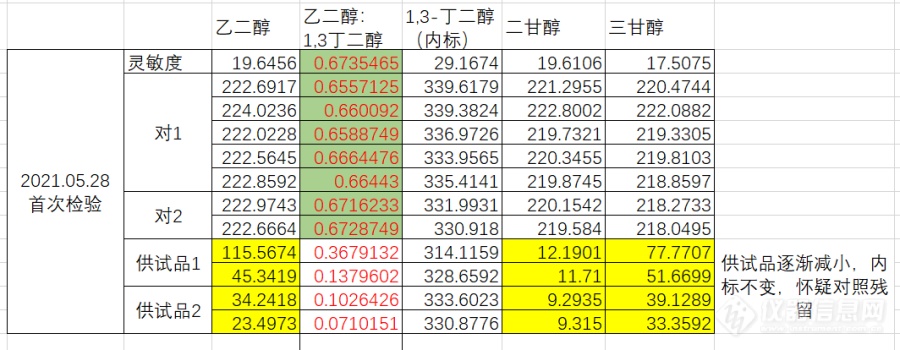
在进行聚山梨酯80Ⅱ 乙二醇、二甘醇和三甘醇检测过程中,发现对照品溶液重复性相当不错,但是供试品中目标峰逐渐减小,见如下数据:[img=第一次实验时实验数据,690,268]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031131180784_3076_3356999_3.png!w690x268.jpg[/img]当时出现这种情况,便怀疑是对照品残留到了供试品中,然后减少了进样针数,实验数据如下:[img=减少对照进样次数,690,147]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031133386103_879_3356999_3.png!w690x147.jpg[/img]供试品确实小了很多,而且也没有逐渐减小的情况,当时基本确定是对照品残留,因此后续实验调整了进样顺序,首先进供试品,但是又出现跟首次检验一样的情况,供试品目标峰面积逐渐减小,而后面的对照重复性也是相当不错做到此时怀疑是色谱柱问题,因此新采购了色谱柱,但供试品逐渐减小的问题仍然存在,后续又按照空白2针,供试品2针,空白2针,供试品2针的顺序进样,数据如下:[img=只进供试品和空白,690,267]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031139383447_5262_3356999_3.png!w690x267.jpg[/img]令本人百思不得其解,请各位分析解答,谢谢~最后附上检验方法(仪器为安捷伦7890B,色谱柱为安捷伦VF-17ms,砂芯衬管)[img=,690,596]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143388912_5805_3356999_3.png!w690x596.jpg[/img][img=,690,368]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143564333_3950_3356999_3.png!w690x368.jpg[/img]
最近在开发一个原料药的乙二醇溶剂残留方法时,遇到一个问题,乙二醇在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中保留时间不一致,容易峰飘,时间相差0.1min中,我们产品是盐酸舒托必利,[img=,679,260]https://ng1.17img.cn/bbsfiles/images/2020/06/202006121037129934_4992_3176442_3.png!w679x260.jpg[/img]是一个盐酸盐,自动进样基质效应很强,所以样品前处理,需要加入氢氧化钠溶液,空白,对照,加标均同法处理,产品在水,甲醇,DMF,DMSO中溶解,开发方法选择用DMF做稀释剂,具体方法如下:见图,后续尝试过,把稀释剂换成水,甲醇,DMSO,依然对照中的乙二醇和加标中的乙二醇峰叠加图中,保留时间还是不能完全重叠,相差0.1min,也尝试过换色谱柱,DB-WAX、DB-624,情况还是没有改变,依然峰飘,一直解决不了这个问题,方法无法进行转移至QC,目前已经在做液相柱前衍生检测,各位老师能否帮忙分析一下,求指导。[img=,591,430]https://ng1.17img.cn/bbsfiles/images/2020/06/202006121036532031_7719_3176442_3.png!w591x430.jpg[/img]
各位师傅,小弟想知道乙二醇不同温度下的密度对照表。望知道的告知一下,谢谢
[color=#333333]乙二醇,丙二醇和二甲基是食品添加剂吗[/color]
供试品溶液制备:取本品1g,精密称定,置顶空瓶中,精密加入超纯水1.0ml, 密封,摇匀,作为供试品溶液。环氧乙烷对照贮备液制备:量取环氧乙烷300μl(相当于0.25g环氧乙烷),置含50ml经过滤处理的聚乙二醇400(以60℃,1.5~2.5kPa旋转蒸发6小时,除去挥发性成分)的100ml量瓶中,加入相同溶剂稀释至刻度,摇匀,作为环氧乙烷对照品贮备液。环氧乙烷对照品溶液制备:精密称取1g冷的环氧乙烷对照品贮备液,置含40ml经过处理的聚乙二醇400的50ml量瓶中,加相同溶剂稀释至刻度。精密称取10g,置含30ml水的50ml量瓶中,加水稀释至刻度。精密量取10ml,置50ml量瓶中,加水稀释至刻度,摇匀,作为环氧乙烷对照品溶液。二氧六环对照品溶液制备:取二氧六环适量,精密称定,用水制成每1ml中含0.1mg的溶液,作为二氧六环对照品溶液。混合对照品溶液制备:精密称取本品1g,置顶空瓶中,精密加入0.5ml环氧乙烷对照品溶液及0.5ml二氧六环对照品溶液,密封,摇匀,作为对照品溶液。系统适用性试验溶液制备:量取0.5ml环氧乙烷对照品溶液置顶空瓶中,加入新鲜配制的0.001%乙醛溶液0.1ml及二氧六环对照品溶液0.1ml,密封,摇匀,作为系统适用性试验溶液。试验条件:照气相色谱法试验,以聚二甲基硅氧烷为固定液,起始温度为35℃,维持5分钟,以每分钟5℃的速率升温至180℃,然后以每分钟30℃的速率升温至230℃,维持5分钟(可根据具体情况调整)。进样口温度150℃,检测器温度250℃,顶空瓶温度70℃,平衡时间为45分钟。取系统适用性试验溶液顶空进样,调节检测器灵敏度使环氧乙烷峰和乙醛峰的峰高约为满量程的15%,乙醛峰和环氧乙烷峰之间的分离度不小于2.0,二氧六环峰高应为基线噪音的5倍以上,分别取供试品溶液及对照品溶液顶空进样,重复进样至少3次。环氧乙烷峰面积的相对标准偏差应不得过15%,二氧六环峰面积的相对标准偏差应不得过10%,按标准加入法计算,环氧乙烷不得过0.0001%,二氧六环不得过0.001%。问题1:“相同溶剂稀释至刻度“是指水吗?问题2:求组计算过程或者指点一下,多谢。实在搞不清楚标准加入法的计算过程,一些人说要扣什么,已经糊涂了,灰常感谢。好人好报
环氧乙烷和二氧六环 取本品1g,精密称定,置顶空瓶中,精密加入超纯水1.0ml,密封,摇匀,作为供试品溶 液。量取环氧乙烷300uL(相当于0.25g环氧乙烷),置含 50ml经过处理的聚乙二醇400(以60℃,1.5~2. 5kPa旋转蒸发6小时,除去挥发性成分)的100ml量瓶中,加入相同溶剂稀释至刻度,摇匀,作为环氧乙烷对照品贮备液,精密称取lg冷的环氧乙烷对照品贮备液,置含40ml经过处理的聚乙二醇400的50ml量瓶中,加相同溶剂稀释至刻度。精密称取10g,置含30ml水的50ml量瓶中,加水稀释至刻 度。精密量取10ml,置50ml量瓶中,加水稀释至刻度,摇匀,作为环氧乙烷对照品溶液。取二氧六环适量,精密称定,用水制成每1ml中含0.1mg的溶液,作为二氧六环对照品溶液。精密称取本品lg,置顶空瓶中,精密加入0.5ml 环氧乙烷对照品溶液及0.5ml 二氧六环对照品溶液,密封, 摇匀,作为对照品溶液。量取0.5ml环氧乙烷对照品溶液置 顶空瓶中,加入新鲜配制的0.001%乙醛溶液0.1ml及二氧六环对照品溶液0.1ml,密封,摇匀,作为系统适用性试验溶液,照[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](通则0521)试验,以聚二甲基硅氧烷为固定液,起始温度为35℃,维持5钟,以每分钟5℃的 速率升温至180℃,然后以每分钟30℃的速率升温至 230℃,维持5分钟(可根据具体情况调整)。进样口温度为 150℃,检测器温度为250℃,顶空平衡温度为70℃,平衡时间为45分钟。取系统适用性试验溶液顶空进样,调节检测器灵敏度使环氧乙烷峰和乙醛峰的峰高约为满量程的 15%,乙醛峰和环氧乙烷峰之间的分离度不小于2.0,二氧六环峰高应为基线噪音的5倍以上,分别取供试品溶液及对照品溶液顶空进样,重复进样至少3次。环氧乙烷峰面积的相对标准偏差应不得过15%,二氧六环峰面积的相对标准偏差应不得过10%,按标准加入法计算,含环氧乙烷不得过0.0001%, 含二氧六环不得过0.001%。
各位有没有做药品乙二醇残留的,求乙二醇的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法!
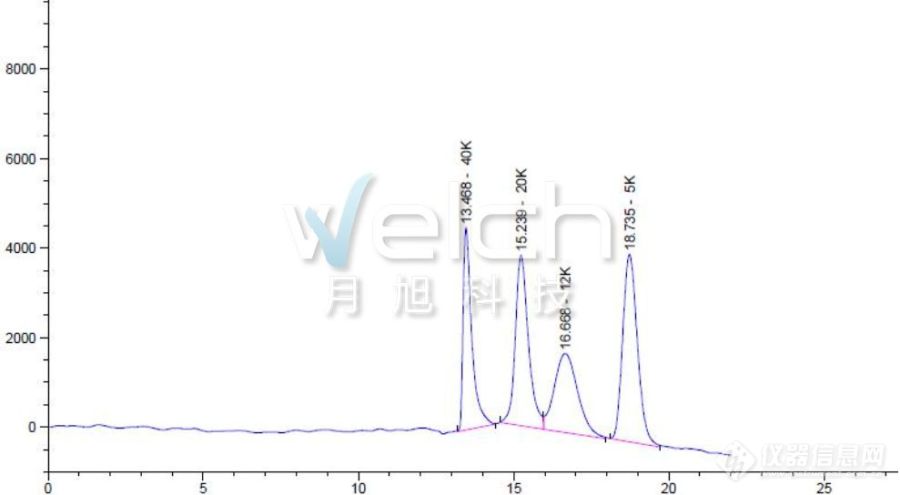
[b][/b][align=center][b]新增项目[/b][/align]最近在浏览国家药典委员会官网的时候,惊奇的发现聚乙二醇300,400,1000……全部的聚乙二醇品种药典方法都要修订了!在浏览了公示方法,发现原来是新增了一个项目,从2020版药典开始要标明重均分子量及分子量分布系数的标示值(按所附测定方法测定),那各位小伙伴了解分子量及分子量分布测定方法吗?这里就给大家详细讲讲。[b]2020版中国药典征求意见稿之分子量及分子量分布测定方法[/b]分别称取聚乙二醇600、聚乙二醇1000、聚乙二醇4000、聚乙二醇7000、聚乙二醇10000分子量对照品适量,加流动相溶解并稀释制成每1ml中约含2mg的溶液作为对照品溶液。称取样品适量,加流动相溶解并稀释制成每1ml中约含2mg的溶液作为供试品溶液。照分子排阻色谱法(通则0514)测定,采用适宜分离范围的凝胶色谱柱,以0.1mol/L硝酸钠溶液(含0.02%抑菌剂)为流动相,示差折光检测器;检测器温度35℃,柱温35℃,取对照品溶液各100μl注入液相色谱仪,记录色谱图,由GPC软件计算回归方程,线性相关系数R应不得小于0.99。取供试品溶液100μl,同法测定,根据回归方程计算供试品的重均分子量及分子量分布。供试品的重均分子量应为标示值的90%~110%,分布系数应为产品标示值的90%~110%。[b]什么是分子排阻色谱法[/b]分子排阻色谱法是根据待测组分的分子大小进行分离的一种液相色谱技术。分子排阻色谱法的分离原理为凝胶色谱柱的分子筛机制。色谱柱多以亲水硅胶、凝胶或经过修饰的凝胶如葡聚糖凝胶(Sephadex)和琼脂糖凝胶(Sepharose)等为填充剂,这些填充剂表面分布着不同孔径尺寸的孔,药物分子进入色谱柱后,它们中的不同组分按其分子大小进入相应的孔内,大于所有孔径的分子不能进入填充剂颗粒内部,在色谱过程中不被保留,最早被流动相洗脱至柱外,表现为保留时间较短;小于所有孔径的分子能自由进入填充剂表面的所有孔径,在色谱柱中滞留时间较长,表现为保留时间较长;其余分子则按分子大小依次被洗脱。[b]分子排阻色谱法应用案例[/b]月旭科技Xtimate SEC色谱柱是硅胶基质的分子排阻色谱柱,其色谱填料为高纯度、具有良好稳定性的硅胶微球表面键合亲水性聚合物。月旭科技采用特殊的表面修饰技术,确保了该填料具有良好的稳定性和批次重现性。Xtimate SEC色谱填料采用独特的化学键合技术,在硅胶表面键合了亲水性聚合物以及亲水性二醇基官能团,双重键合机制使水溶性高分子聚合物蛋白、生物酶、多肽等生物样品的非特异性吸附极小,因而可广泛应用于水溶性聚合物及生物大分子的分离和测定。月旭科技采用Xtimate SEC-300 (7.8*300mm,5μm)两根色谱柱串联的方式成功分离检测聚乙二醇40k,20k,12k,5k含量。[b]色谱柱:[/b]月旭Xtimate SEC-300 (7.8*300mm,5μm)两根色谱柱串联[b]流动相:[/b]高纯水[b]检测波长:[/b]示差检测器[b]柱温:[/b]柱温40℃,检测器40℃[b]流速:[/b]1.0ml/min[b]进样量:[/b]20μl[align=center][img=,690,379]https://ng1.17img.cn/bbsfiles/images/2019/10/201910160944591635_8915_932_3.jpg!w690x379.jpg[/img][/align][align=center][img=,414,40]https://ng1.17img.cn/bbsfiles/images/2019/10/201910160946105076_5570_932_3.png!w414x40.jpg[/img][/align]各位小伙伴想了解更多有关分子排阻色谱法的相关信息,请咨询月旭当地销售人员或拨打400-810-6969垂询。
[b][font=宋体][color=red]乙二醇、二甘醇和三甘醇[/color][/font][color=red] [/color][/b][font=宋体][color=red]供试品溶液:取本品[/color][/font][color=red]4g[/color][font=宋体][color=red],精密称定,置[/color][/font][color=red]100ml[/color][font=宋体][color=red]量瓶中,精密加入内标溶液(取[/color][/font][color=red]1[/color][font=宋体][color=red],[/color][/font][color=red]3-[/color][font=宋体][color=red]丁二醇适量,用无水乙醇稀释制成每[/color][/font][color=red]1ml[/color][font=宋体][color=red]中约含[/color][/font][color=red]4mg[/color][font=宋体][color=red]的溶液)[/color][/font][color=red]1.0ml[/color][font=宋体][color=red],加无水乙醇稀释至刻度,摇匀,作为供试品溶液。[/color][/font][font=宋体][color=red]对照品溶液:取乙二醇、二甘醇和三甘醇对照品适量,精密称定,加无水乙醇稀释配制成每[/color][/font][color=red]1ml[/color][font=宋体][color=red]含乙二醇、二甘醇、三甘醇[/color][/font][color=red]4mg[/color][font=宋体][color=red]的溶液,作为对照品贮备液;精密量取对照品贮备液[/color][/font][color=red]1.0ml[/color][font=宋体][color=red]与内标溶液[/color][/font][color=red]1.0ml[/color][font=宋体][color=red],置[/color][/font][color=red]100ml[/color][font=宋体][color=red]量瓶中,加无水乙醇稀释至刻度,摇匀,作为对照品溶液。[/color][/font][font=宋体][color=red]灵敏度溶液:精密量取对照品溶液[/color][/font][color=red]1.0ml[/color][font=宋体][color=red],置[/color][/font][color=red]l0ml[/color][font=宋体][color=red]量瓶中,加无水乙醇稀释至刻度,摇匀,作为灵敏度溶液。[/color][/font][font=宋体][color=red]用的柱子是DB-17([/color][/font][color=red]50%[/color][font=宋体][color=red]苯基[/color][/font][color=red]-50%[/color][font=宋体][color=red]甲基聚硅氧烷为固定液([/color][/font][color=red]30m×0.53mm[/color][font=宋体][color=red],[/color][/font][color=red]1.0μm[/color][color=red])[font=宋体][color=#ff0000])[/color][/font],起始温度为[/color][color=red]40[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red],以每分钟[/color][/font][color=red]10[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red]的速率升温至[/color][/font][color=red]60[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red],维持[/color][/font][color=red]5[/color][font=宋体][color=red]分钟后,以每分钟[/color][/font][color=red]5[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red]的速率升温至[/color][/font][color=red]110[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red],维持[/color][/font][color=red]5[/color][font=宋体][color=red]分钟,再以每分钟[/color][/font][color=red]5[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red]的速率升温至[/color][/font][color=red]170[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red],维持[/color][/font][color=red]5[/color][font=宋体][color=red]分钟,再以每分钟[/color][/font][color=red]35[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red]的速率升温至[/color][/font][color=red]280[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red],[/color][/font][font=宋体][color=red]维持[/color][/font][color=red]30[/color][font=宋体][color=red]分钟。进样口温度为[/color][/font][color=red]270[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red],氢火焰离子化检测器温度为[/color][/font][color=red]290[/color][font=宋体][color=red]℃[/color][/font][font=宋体][color=red]。[/color][/font][font=宋体][color=red]精密量取灵敏度溶液[/color][/font][color=red]1μl[/color][font=宋体][color=red],进样,调节检测灵敏度使乙二醇、二甘醇和三甘醇峰的信噪比均大于[/color][/font][color=red]10[/color][font=宋体][color=red]。[/color][/font][font=宋体][color=red]问题是这个灵敏度溶液中乙二醇一直不出峰,二甘醇和三甘醇都有峰,对照溶液三个对照都有峰,想问问这种情况该怎么解决?[/color][/font]
1.如何分析丙烯酸中的乙二醛,水中乙二醇? 乙二醛会导致丙烯酸聚合,应越少越好。乙二醇为抗冻剂,检查工业水中的乙二醇含量可以确认冷冻水管是否破裂,滲漏。1.曾以GC,LC,UV,GC-MASS进行尝试,均定性不出来。 GC上曾尝试DB-WAX,HP-5,FFAP等不同的管住进行分析。 LC的管住为SB-C8,加入不同浓度的标准品,峰形却几乎不变。波长扫描后,换波长也没用。 请教各位有什么高招进行分析。
最近发现聚乙二醇400、乙二醇的新用途,但不想给人检测出来,请教各位高手,要加什么东西,才能不给人轻易检测出来。急!!!!
请教聚乙二醇、二乙二醇的分析方法或标准,哪位知道的大侠告诉下哦,谢谢。。。
大家好,谁在乙二醇项目上呢,本人想求购一本乙二醇化验室相关资料,
大家用HP-5的柱子分析过高纯度二乙二醇中微量一乙二醇和三乙二醇的么?
外标法做乙二醇原料,色谱图中乙二醇为50%,单独测定的水为30%,请问原料中的乙二醇含量为多少?
在论坛看到了有关的资料,但都是用聚乙二醇柱子或非极性柱子分的,我们现在只有DM1701的柱子,好像是中等极性的,试了很多条件都分不开。按理说既然不管根据极性还是沸点都能分开,且都是乙二醇先出峰的话,用1701的应该也能分开阿(是本人想当然的,见笑了)。请问如果我只能用1701的柱子的话,还需要怎么优化条件才能分开呢?
[em61] 各位论友,帮帮忙!我现有一样品,其中主要分析乙二醇与苯甲醇,现求助大家帮我确定以下色谱柱以及分析条件? 非常感谢!
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析乙醇和乙二醇 乙二醇含量约0.5% 连续6次手动进样乙二醇峰面积RSD%是5.246 我想继续改善分析的重现性及准确性 所以想选用内标曲线法 请问我选取何种内标物合适?[/color]
甘油有关物质,安捷伦[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]色谱柱用DB-624:30 0.53 3.0大口径色谱柱流速4ml/min,分流比10:1,恒定流量模式,其他条件药典规定。柱效,分离度挺好。但是进完系统适用性后,走对照时,乙二醇和丙二醇峰面积成递增状态导致RSD不行。[img]https://ng1.17img.cn/bbsfiles/images/2023/06/202306282316101695_2371_6077845_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/06/202306282316101715_6855_6077845_3.png[/img]
1.如何分析丙烯酸中的乙二醛,水中乙二醇? 乙二醛会导致丙烯酸聚合,应越少越好。乙二醇为抗冻剂,检查工业水中的乙二醇含量可以确认冷冻水管是否破裂,滲漏。1.曾以GC,LC,UV,GC-MASS进行尝试,均定性不出来。 GC上曾尝试DB-WAX,HP-5,FFAP等不同的管住进行分析。 LC的管住为SB-C8,加入不同浓度的标准品,峰形却几乎不变。波长扫描后,换波长也没用。请教各位有什么高招进行分析。
请问下有使用USP药典方法检测聚乙二醇中乙二醇和二甘醇过吗?色谱柱为Chromosorb WNAW 12%山梨醇,1/8''*2.0mm*1.5m,柱温140℃,在考察该方法的时候发现第一次实验空白无干扰,第二次实验空白在乙二醇和二甘醇出峰处出峰,请问下是因为前一次做的样品残留在填充柱中导致空白出峰的吗?[img]https://ng1.17img.cn/bbsfiles/images/2019/11/201911201543024359_9780_3860760_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/11/201911201543002954_9705_3860760_3.png[/img]
最近购入二乙二醇试剂,使用时用30m的PEG柱子分析了一下纯度99.5,其中可能含有乙二醇和三乙二醇,可是再次测定时为什么这两个出峰的量不一致了呢?是使用柱子时脱掉柱子的问题吗或者其他的什么问题,望各位老师赐教,谢谢。。。。
2015版药典四部测聚乙二醇4000中环氧乙烷和二氧六环实验,对照品为什么要先用聚乙二醇400溶解,最后用水稀释,供试品都是用水溶解的,能不能都用水溶解啊
聚乙二醇[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]残留做二甘醇和乙二醇:药典用注射进样,由于,聚乙二醇杂峰很多,做不出来,我想改用顶空进样,可二甘醇和乙二醇的沸点很高,不知道可不可行?谢谢
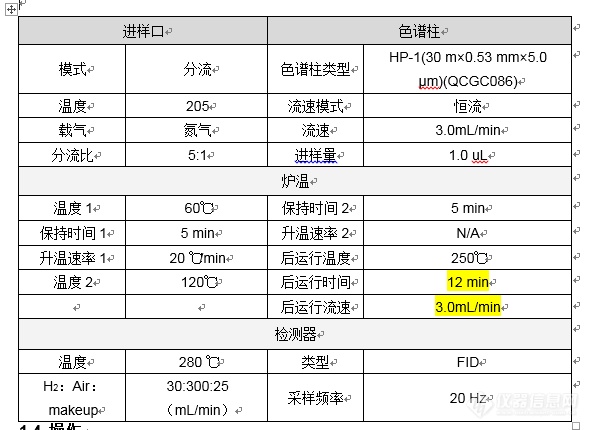
乙二醇测试条件及谱图







 我要推广仪器
我要推广仪器
 下载APP
下载APP