李灵军与叶慧团队合作成果:生物素硫醇标签辅助质谱法对蛋白质瓜氨酸化进行全局分析
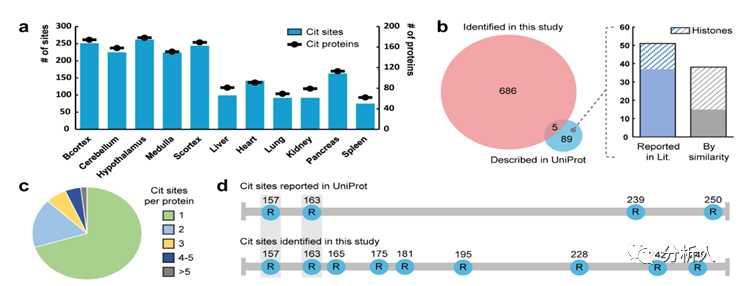
瓜氨酸化是影响蛋白质结构和功能的关键的翻译后修饰。尽管它与各种生物过程和疾病发病紧密相关,但由于缺乏有效的方法来富集、检测和定位该翻译后修饰,其潜在机制仍然知之甚少。近期,威斯康星大学麦迪逊分校李灵军教授课题组报道了生物素硫醇标签的设计和开发,该标签能够通过质谱法对瓜氨酸化进行衍生化、富集来实现可靠的鉴定。作者对小鼠组织的瓜氨酸化蛋白质组进行了全局分析并且从432种瓜氨酸化蛋白质中识别出691个修饰位点,这是迄今为止最大的瓜氨酸化数据集。作者发现并阐述了这个翻译后修饰的新的分布和功能并且表示该方法有希望为进一步破译瓜氨酸化的生理和病理作用奠定基础。这项工作以“Enabling Global Analysis Of Protein Citrullination Via Biotin Thiol Tag-Assisted Mass Spectrometry”为题发表在国际化学权威杂志Analytical Chemistry上 (https://doi.org/10.1021/acs.analchem.2c03844),文章作者为Yatao Shi#, Zihui Li#, Bin Wang#,Xudong Shi , Hui Ye, Daniel G. Delafield, Langlang Lv, Zhengqing Ye, Zhengwei Chen, Fengfei Ma,Lingjun Li*。此外,李灵军教授课题组进一步拓展了此方法的实用性。作者通过应用二甲基化亮氨酸(DiLeu)等重标记策略第一次实现了瓜氨酸化的高通量定量研究,并利用这一方法揭示了瓜氨酸化在人体细胞DNA损伤及修复过程中的重要作用。相关成果以“12-Plex DiLeu Isobaric Labeling Enabled High-Throughput Investigation of Citrullination Alterations in the DNA Damage Response”为题同样发表在Analytical Chemistry上(https://doi.org/10.1021/acs.analchem.1c04073),文章作者为Zihui Li, Bin Wang, Qinying Yu, Yatao Shi, Lingjun Li*。 研究的主要内容 作者设计了一种生物素硫醇标签,它可以很容易的以低成本合成并且可以与瓜氨酸残基和2,3-丁二酮发生特异性反应(图 1a)。这种衍生化不仅增加了质量转移以允许更可靠的鉴定,而且还引入了生物素部分,使修饰分子的后续富集成为可能。该生物素硫醇标签设计具有紧凑的结构,在高能碰撞解离 (HCD) 期间仅产生两个碎片/诊断离子(图 1b)。 因此,肽主链可以保持良好的裂解效率,并在 HCD 或电子转移解离 (ETD) 期间分别产生丰富的b/y或c/z离子系列。在 HCD(图 1c)、ETD或电子转移/高能碰撞解离(EThcD)碎裂下,衍生化肽标准品的序列收集质谱图几乎完全覆盖相应的肽序列。实验结果表明生物素硫醇标签衍生的瓜氨酸化肽可以产生用于解析及标注的高质量的串联质谱图,并且与各种裂解技术相结合时可以提高瓜氨酸化位点的识别可信度。 图1|用于瓜氨酸化分析的生物素硫醇标签设计。a,使用生物素硫醇标签和 2,3-丁二酮对瓜氨酸肽进行衍生化。 b,HCD、ETD 或 EThcD 片段化后生物素硫醇标签衍生的瓜氨酸化肽的片段化位点。c,HCD 裂解后生物素硫醇标签衍生的瓜氨酸肽标准品 SAVRACitSSVPGVR 的串联质谱图。 在接下来的实验中作者使用该生物素硫醇标签和基于质谱的自下而上的蛋白质组学方法对瓜氨酸化进行分析(图2a)。作者在体外利用 PAD(一种可以催化瓜氨酸化的酶)催化的人组蛋白 H3 蛋白来验证这个过程。作为未被PAD催化的阴性对照,未发现组蛋白的肽段被鉴定为瓜氨酸化,证明了生物素标签反应的高特异性(图 2b)。在体外 PAD 处理后,作者 发现许多精氨酸残基被催化为瓜氨酸,并且大量的位点被高可信度的鉴定为瓜氨酸化位点(图 2c),进一步表明该方法的高效性。在 HCD 碎裂后,其产生了一系列丰富的 b/y 离子,可以帮助准确的表征在同一肽段上单个(图 2d)以及多个(图 2e)瓜氨酸化位点。 图2|使用生物素硫醇标签进行体外瓜氨酸化分析。a,使用生物素硫醇标签进行蛋白质瓜氨酸化分析的实验工作流程。b、c,在体外 PAD 处理之前 (b) 和之后 (c) 组蛋白 H3 蛋白的瓜氨酸化分析。 已识别的瓜氨酸化位点在序列中以蓝色字母突出显示。 序列下方的红色矩形表示鉴定的瓜氨酸化肽,而瓜氨酸化位点以蓝色显示。 d,PAD处理的组蛋白 H3 (R64Cit) 的已鉴定瓜氨酸化肽的串联质谱图示例。 e,PAD 处理的组蛋白 H3 的同一肽上鉴定的两个瓜氨酸化位点(R70Cit 和 R73Cit)的串联质谱图示例。 接下来,作者们尝试利用所开发的方法对复杂的生物样本中的瓜氨酸化进行全局分析,并希望能够以此提供阐明生物体中瓜氨酸化调节机制的依据。首先,作者对小鼠的六个身体器官和五个大脑区域进行了深入的瓜氨酸组分析,生成了第一个小鼠瓜氨酸组组织特异性数据库。作者从432种瓜氨酸化蛋白质中以高置信度的方式鉴定了691个瓜氨酸化位点(图 3a)。更重要的是,这些蛋白质中约有 60% 未曾在UniProt 数据库检索并被报道,这一结果极大地扩展了对瓜氨酸化以及这些底物蛋白质如何受到瓜氨酸化影响的理解。作者发现结果中与 UniProt 数据库的已知的瓜氨酸位点重叠部分较少(图 3b),这可能是因为 UniProt 中描述的近 40% 的瓜氨酸化位点是基于相似性外推理论而没有实际的实验证据。此外,许多报道的位点位于组蛋白上,尤其是蛋白质末端,可能会逃过自下而上质谱策略的检测(图 3b)。图 3c 展示了单位点瓜氨酸化和多位点瓜氨酸化蛋白质分布情况,其中 70% 的已鉴定蛋白质仅有一个瓜氨酸化位点被检测到。 这个新发现的瓜氨酸化蛋白质组为推测瓜氨酸化的调控机制提供了宝贵的资源。例如,作者在髓鞘碱性蛋白(MBP)上鉴定到了九个瓜氨酸化位点,而在 UniProt 数据库中只有四个(图3d)。作者的结果提供了高质量的串联质谱图,不仅证实了已知修饰位点的存在(图3e),而且还高可信度的识别了未知的位点(图 3f)。然后作者进行了瓜氨酸化肽段的序列分析,发现在鉴定的瓜氨酸化位点两侧并没有高度保守的氨基酸序列模式(图3g),但是谷氨酸残基更频繁地出现在瓜氨酸的N末端侧附近。这与Fert-Bober 等人报道的小鼠瓜氨酸组分析结论一致。另一方面,Tanikawa 等人发现在人体组织和血浆中大约五分之一的 PAD4 底物含有 RG/RGG 基序。同样,Lee 等人及相关研究人员观察到天冬氨酸和甘氨酸残基在瓜氨酸化位点出现频率偏高。值得注意的是,这些研究使用了不同的人源细胞系或组织,因此作者的结果可能表明在不同物种之间瓜氨酸化位点周围的序列模式是不同的。为了更好地辨别瓜氨酸化蛋白质所涉及的功能,作者展示了基因本体论(GO)富集分析的热图,其显示了二十个最显著富集的细胞成分(图3h)以及KEGG途径(图3i)。作者发现小鼠大脑组织和身体器官之间存在明显差异,而瓜氨酸蛋白更多地参与大脑功能。具体来说瓜氨酸化蛋白质集中在轴突、髓鞘、核周体和突触中,因此在中枢神经系统中可能发挥着重要的作用。 图3|不同小鼠组织的大规模瓜氨酸组分析。a,不同小鼠组织中已鉴定的瓜氨酸化蛋白和瓜氨酸化位点的数量。 b,本研究中鉴定的瓜氨酸化位点与 UniProt 数据库中报告的位点比较。 c,每个鉴定的瓜氨酸化蛋白质的瓜氨酸化位点数量分布。d,本研究中确定的瓜氨酸化位点与 UniProt 数据库中关于髓鞘碱性蛋白的瓜氨酸化位点的比较。e、f,在髓磷脂碱性蛋白 R157Cit (e) 和 R228Cit (f) 上鉴定的两个瓜氨酸化位点的示例串联质谱图。g,鉴定的瓜氨酸化肽的序列。瓜氨酸化位点位于中间的“0”位置。字母的高度表示每个氨基酸在特定位置的相对频率。 h,i,使用 Metascape 生成的热图显示不同小鼠组织中显着丰富的(p 值 0.01)细胞成分 (h) (KEGG) 通路 (i)。 为了进一步拓展该方法的实用性,作者应用了二甲基化亮氨酸(DiLeu)等重标记策略,第一次实现了对瓜氨酸化进行高通量的定量研究。作者首先使用瓜氨酸化标准肽段进行测试,证明在优化反应条件下DiLeu标记和生物素硫醇标记反应可以分步进行而不互相干扰(图 4B,4C)。同时,将标准肽段按照已知比例进行4-plex DiLeu标记并混合,再进行生物素硫醇标记和瓜氨酸化分析,结果显示了非常好的定量准确性(图5)。作者进一步优化了运用该方法在复杂生物样品中进行定量分析的实验方法,并且证明此方法依然可以实现极佳的定量准确度和精确度(图6)。 图4|瓜氨酸化标准肽段测试DiLeu标记和生物素硫醇标记分步反应的特异性和效率 图5|瓜氨酸化标准肽段测试DiLeu标记和生物素硫醇标记定量分析的准确性 图6|复杂生物样品测试DiLeu标记和生物素硫醇标记定量分析的准确度和精确度 作者接下来应用该方法对DNA损伤中瓜氨酸化的作用进行了研究。作者在MCF7细胞中用三种方法造成了DNA损伤,并定量分析了蛋白质瓜氨酸化的变化。作者一共鉴定到63种瓜氨酸化蛋白以及其包含的78个瓜氨酸化位点,并发现三个实验组中的瓜氨酸化表达相比于对照组呈现出非常不同的趋势(图7A),这一结果表明瓜氨酸化在不同类型的DNA损伤模型中具有差异性的作用。通过对实验组中显著变化的瓜氨酸化蛋白进行生物过程网络分析,作者发现瓜氨酸化主要对DNA代谢,蛋白结构变化,翻译以及DNA修复等过程进行调控(图 7B,7C)。该实验结果表明蛋白瓜氨酸化对DNA损伤以及相关发病机理具有非常重要的作用。 图7|高通量定量分析研究瓜氨酸化在DNA损伤中的变化及作用(来源:Anal. Chem.) 小结 本文章介绍了一种生物素硫醇标签的设计和开发,该标签可与瓜氨酸化肽段发生特异性反应并极大地提高了瓜氨酸化的富集和检测效率。在使用标准肽和重组蛋白证明该方法的有效性后,作者进一步优化了从复杂生物样品中检测瓜氨酸化的实验过程。通过此方法对小鼠五个大脑区域和六个身体器官的蛋白质瓜氨酸化进行分析,作者鉴定出432个瓜氨酸化蛋白以及691个瓜氨酸化位点,这是迄今为止最大的数据集。该研究揭示了这种翻译后修饰可能在神经系统中发挥的关键作用,并表明它们在包括呼吸和糖酵解在内的许多代谢过程中也可能发挥着重要作用。总的来说,实验结果表明蛋白质瓜氨酸化在不同组织中具有广泛分布并参与各种生物过程,这扩展了目前对蛋白质瓜氨酸化生理作用的认知和理解。此外,作者进一步拓展了此方法的实用性,通过应用DiLeu等重标记策略第一次实现了瓜氨酸化的高通量定量研究,并利用这一方法揭示了瓜氨酸化在人体细胞DNA损伤及修复过程中的重要作用。更重要的是,该方法可以提供一种普适、简单而强大的检测方法来明确鉴定蛋白质瓜氨酸化,这也将启发和有益于未来对这种翻译后修饰在生理和病理条件下的功能作用的研究。 相关研究成果近期发表在Analytical Chemistry上的两篇文章中, 通过生物素硫醇标签辅助质谱法对蛋白质瓜氨酸化进行全局分析文章的共同第一作者是威斯康星大学麦迪逊分校博士生石亚涛,李子辉,王斌,并与中国药科大学叶慧教授课题组合作 应用二甲基化亮氨酸等重标记策略进行蛋白质瓜氨酸化高通量定量研究文章的第一作者是威斯康星大学麦迪逊分校博士生李子辉,两篇文章通讯作者为李灵军教授。更多关于李灵军教授研究团队的最新研究进展欢迎登陆课题组网站:https://www.lilabs.org/
























 我要推广仪器
我要推广仪器
 下载APP
下载APP