
内标跟着文献选的L-正亮氨酸,实验室正好有L-亮氨酸请问能用吗[img=,300,400]https://ng1.17img.cn/bbsfiles/images/2022/11/202211221916333293_727_5869599_3.jpg!w690x920.jpg[/img]
我的产物是L-叔亮氨酸,但高效用的标品是其盐酸盐,请问会有什么影响?[em0808]
哪位老师知道L-异亮氨酸的测定,请告知。谢谢!
[color=#444444]有没有哪位大侠做过叔亮氨酸的液相检测,文献里面查到1.用手性色谱柱,以2mM硫酸铜、5%异丙醇做流动相,流速1mL/min,254nm检测;2.同样是手性柱,用硫酸铜做流动相,检测波长为220nm。3.用C18柱,以0.25%磷酸二氢铵:甲醇=100: 5,检测波长205nm。4.另外有用OPA衍生后再检测的,检测波长340nm。由于目前没有标品,不知道叔亮氨酸的最大吸收波长在什么位置,叔亮氨酸上没有共轭结构,254nm检测是不是不靠谱啊?求有相关经验的大大指条明路[/color][img=,absmiddle]http://muchongimg.xmcimg.com/data/emuch_bbs_images/smilies/wink.gif[/img][img=,absmiddle]http://muchongimg.xmcimg.com/data/emuch_bbs_images/smilies/wink.gif[/img][color=#444444]另外听说手性柱金贵的很,求指教平时使用的注意点[/color][img=,absmiddle]http://muchongimg.xmcimg.com/data/emuch_bbs_images/smilies/biggrin.gif[/img]
大家好,我们在进行蛋白质修饰鉴定过程中,发现有异亮氨酸甲基化的修饰(采用二级CID碎裂模式),分析软件(BioPharmaView)中给定的修饰中也含有异亮氨酸,为了确定甲基化修饰的机理,我们推测,甲基化修饰在了异亮氨酸形成的肽键N上,对此我们使用etHCD碎裂模式进行二级碎裂,结果显示,甲基化并非修饰在肽键N上,我们查询文献并没有相关的报道,想问下各位大神,有知道蛋白中异亮氨酸发生甲基化是发生在哪个位置么?如果有文献支持就更好了。
我用GC MS 测20种氨基酸,MSTFA衍生,不加溶剂,HP 5-MS柱,70度,1min到5度/min,300度,得到的脯氨酸和异亮氨酸是一个峰,降低浓度也分不开,做SIM也分不开,请问谁遇到过这种情况?如何解决?
如题!求助亮氨酸快速检测方法!现有硅胶板层析法,速度较慢,耗时1h,不知谁有好的方法?
[color=#444444]用OPA柱前衍生反向高效液相色谱测定L-叔亮氨酸的色谱图中出现了两个面积差不多的峰是什么原因?叔亮氨酸是手性氨基酸,流动相A用的是20mmol/ L 乙酸钠缓冲液,用1 %稀乙酸调p H 至71 2 流动相B ∶乙腈和甲醇混合液(1 ∶1),洗脱程序是等度洗脱,A:B是3:2. 求解释!!![/color]

先简单 介绍——————做氨基酸 检测想了解详细资料,请自己到迪马科技官网自行下载http://simg.instrument.com.cn/bbs/images/brow/em09510.gifPITC柱前衍生法18种天然氨基酸分析(异硫氰酸苯酯柱前衍生法)——序列号: D0241 适用范围 该方法适用于氨基酸注射液、动植物性食品和饲料中 Asp(天冬氨酸)、Glu(谷氨酸)、Ser(丝氨酸)、Gly(甘氨酸)、His(组氨酸)、Arg(精氨酸)、Thr(苏氨酸)、Ala(丙氨酸)、Pro(脯氨酸)、Tyr(酪氨酸)、Val(缬氨酸)、Met(蛋氨酸)、Cys(胱氨酸)、Ile(异亮氨酸)、Leu(亮氨酸)、Phe(苯丙氨酸)、Trp(色氨酸)、 Lys(赖氨酸)等 18种天然氨基酸的检测http://ng1.17img.cn/bbsfiles/images/2012/03/201203131711_354396_2019107_3.jpg2 溶液配制 氨基酸储备液: 称取一定量氨基酸标准品,用 0.1 mol/L HCl水溶液溶解,胱氨酸为0.01 mol/L,酪氨酸为0.02 mol/L,其他氨基酸为 0.05 mol/L 氨基酸使用液: 将储备液用0.1 mol/L HCl水溶液稀释,得到浓度为 0.002 mol/L 的氨基酸单标和混标 内标液: 以正亮氨酸作为内标物。称取一定量正亮氨酸,溶于 0.1 mol/L HCl水溶液,得到 0.02 mol/L 的正亮氨酸内标液 异硫氰酸苯酯溶液: 将 250 μl 异硫氰酸苯酯用乙腈乙腈定容至 10 ml,得到0.2 mol/L 异硫氰酸苯酯溶液 三乙胺溶液: 将1.4 ml三乙胺用乙腈定容至 10 ml,得到1.0 mol/L 三乙胺溶液 标准溶液衍生化 量取 200 µl氨基酸混合标准溶液(每种组分浓度均为 0.002 mol/L),置于 1.5 ml塑料离心管中,准确加入20 μl正亮氨酸内标溶液、100 µl 1 mol/L三乙胺乙腈溶液和100 µl 0.2 mol/L 异硫氰酸苯酯乙腈溶液,混匀,室温反应 1 小时,然后加入正己烷 400 µl,旋紧盖子后剧烈振荡5~10 s,静置分层,取 200 µl下层溶液与 800 µl水混合,0.22 µm 针式过滤器过滤,待分析。注: 通过控制原始样品质量或稀释等方法,使样品溶液中的氨基酸总量不超过0.04 mol/L 或3.0 g/L(两者中取最小值) 只有采用内标法分析时,才需要加入正亮氨酸作为内标物 衍生得到的样品溶液中含有50%的乙腈,这与流动相溶剂体系存在较大差距,因而需要加水稀释,否则会引起峰前沿或分叉迪马科技AAA氨基酸柱子 洗脱条件 http://ng1.17img.cn/bbsfiles/images/2017/01/201701191656_646181_2019107_3.gifhttp://ng1.17img.cn/bbsfiles/images/2011/04/201104221943_290383_2019107_3.gif
求助一下: 今天发现中国药典2005版红外光谱图集中“组氨酸”的对照图谱为什么有两个,一个是光谱号785,另一个是光谱号981。高手回答一下,两者有什么区别? 组氨酸的专论中红外鉴别是要求与981一致,而没有提到785.http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
本人在8月发表的一篇原创中提及”甘氨酸与组氨酸无法分离“的问题,在经过10多天的准备,已有不小的收获,现在分享。摘要 目的: 建立用高效液相色谱法测定人凝血因子VIII中氨基酸含量。方法: 采用6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 为衍生剂,与氨基酸柱前衍生后,用Agilent 1200 高效液相色谱仪,AccQ·Tag C18柱( waters 150 mm ×3. 9 mm,4 μm) ,以水Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液和乙腈进行梯度洗脱,检测波长为248 nm,柱温37 ℃,进样量10μL。结果: 各氨基酸在32 min 内测定完毕,回收率为98.7% ~ 101.5%。RSD 均小于1. 5%。结论: 本法分离度好,快速、简便,可作为产品的质量控制方法。关键词: 6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯; 人凝血因子VIII; 甘氨酸; 衍生物; 梯度洗脱; 高效液相色谱法;氨基酸; 含量测定人凝血因子VIII,本品对缺乏人凝血因子礓所致的凝血机能障碍具有纠正作用,主要用于防治甲型血友病和获得性凝血因子Ⅷ缺乏而致的出血症状及这类病人的手术出血治疗。该药物制备过程中使用了氨基酸( 精氨酸、丙氨酸、甘氨酸、组氨酸、盐酸赖氨酸、脯氨酸 等) 做稳定剂,为了保证药品质量和用药安全,应对其中氨基酸的含量进行控制。该法依据过量的6 - 氨基喹啉基- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 在一定条件和氨基酸形成稳定的衍生产物( 柱前衍生) ,用高效液相色谱法测定衍生产物,根据衍生产物的含量计算人凝血因子中各氨基酸的含量。1 仪器和试药1200 高效液相色谱系统( 美国Agilent 公司) ,配置低压四元梯度泵、1314B 紫外吸收检测器、自动进样器、柱温箱、Chemistations 化学工作站; Sartorius CP225D 电子微量天平( 德国Sartorius 公司) ; SartoriusPB - 21 型pH 计( 德国Sartorius 公司) ; LDZ5 -2 低速自动平衡离心机( 上海医用离心机厂) 等。各标准品均来自于中国食品药品检定研究院2 色谱条件及系统适用性试验色谱柱: Waters AccQ·Tag C18色谱柱( 3. 9 mm ×150 mm) ; 流动相: 水为溶剂D,Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液( A) - 乙腈( B) - 水( D) ,柱温:37 ℃; 检测波长: 248 nm。精密量取对照品溶液与供试品溶液10 μL,分别注入液相色谱仪,记录色谱图32 min。3 溶液制备3. 1 Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液称取三水乙酸钠190. 4 g,加注射用水1000 mL,搅拌,溶解,用稀磷酸将pH 调至5. 2,加入乙二胺四乙酸二钠溶液( 称取乙二胺四乙酸二钠100 mg,加注射用水100 mL,摇匀使其溶解) 10 mL,加入叠氮化钠0. 1 g 及三乙胺23. 7 mL( 17. 2 g) ,用稀磷酸滴定至pH 4. 95,用0. 45 μm 的滤膜过滤,于4 ℃储存,备用( 此条件下可保存6 个月) 。量取该溶液100 mL,加注射用水稀释至1000 mL,混匀,即得Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液。3. 2 对照品储备液混合对照品储备液精密称取各氨基酸对照品适量,置同一100 mL量瓶中,以注射用水溶解并定容至刻度。制成含氨基酸含量均含5. 0 mg·mL - 1 的混合对照品溶液,即得。单个对照品储备液: 精密称取各含氨基酸的各对照品适量,分别置100mL 量瓶中,用注射用水溶解并定容至刻度。制成分别含各氨基酸的单个对照品溶液,即得。3. 3 供试品储备液3. 3. 1 加样回收率试验溶液精密称取各氨基酸各0. 3200,0. 4000,0. 4800 g 和辅料适量,加人凝血因子VIII原液7. 5 mL,肝素钠适量,用1. 0 mol·L - 1 盐酸调pH 至6. 9,加0. 01 mol·L - 1枸橼酸三钠溶液溶解并定容于20 mL。分别制备成16. 0, 20. 0, 24. 0 mg·mL - 1溶液。3. 3. 2 空白溶液 按公司处方,加入辅料的混合物,用注射用水制备各空白溶液3. 4 内标溶液精密称取α - 氨基丁酸( AABA)0. 4 g,加注射用水定容至100 mL。4 氨基酸衍生方法4. 1 精密量取供试品储备液、样品及对照品储备液各1. 0 mL,加1. 5%磺基水杨酸9. 0 mL,混匀静置2 h以上, 3000 r·min - 1离心10 min,留取上清液。4. 2 精密量取“4. 1”项下上清液1. 0 mL( 其中对照品储备液对应上清液分别精密量取0. 06, 0. 4,0. 8,1. 0, 1. 2, 1. 6 mL) ,分别置10 mL 量瓶中,用注射用水定容。制备成供试品溶液、样品溶液及浓度分别为1. 5, 10. 0, 20. 0, 25. 0, 30. 0,40. 0 mg·mL - 1 的对照品溶液。4. 3 精密量取“4. 2”项下溶液各100 μL,分别加注射用水0. 4 mL 及内标溶液20 μL,混匀备用。4. 4 精密量取“4. 3”项下溶液30 μL 放入衍生管中,加硼酸缓冲液( pH 8 ~ 10) 210 μL 涡旋混合,并加入AQC 衍生剂60 μL 涡旋混合15 s,即为各供试品溶液,待用。
阿胶的含量测定的时候,对照品和供试品出的L-羟脯氨酸的理论塔板数都不够是怎么回事啊?就算供试品是处理的问题,但是对照品在处理上不复杂也没有出差错啊!求大师指教!
氨基酸是氨基和羧基的一类有机化合物的通称。生物功能大分子蛋白质的基本组成单位,是构成动物营养所需蛋白质的基本物质。是含有一个碱性氨基和一个酸性羧基的有机化合物。无色晶体,熔点极高,一般在200℃以上。不同的氨基酸其味不同,有的无味,有的味甜,有的味苦。各种氨基酸在水中的溶解度差别很大,并能溶解于稀酸或稀碱中,但不能溶于有机溶剂。近年来,随着技术的发展,检测氨基酸的种类及含量的方法很多,目前测定氨基酸的方法主要有氨基酸分析仪法、高效液相色谱法、毛细管电泳法及液相色谱-质谱联用法等。本文主要是通过高效液相串联质谱法来对16种氨基酸的含量进行测定,方法简单,快速,不用柱前衍生,节省试剂和成本,为保健品中氨基酸的含量测定提供了新方法。1实验部分1.1 仪器和试剂 16种氨基酸的混合标准品,(包括丙氨酸、精氨酸、天冬氨酸、胱氨酸、谷氨酸、组氨酸、亮氨酸、异亮氨酸、赖氨酸、蛋氨酸、苯丙氨酸、脯氨酸、苏氨酸、酪氨酸、缬氨酸、丝氨酸)安捷伦;乙腈(色谱纯)CNW;乙酸铵(色谱纯);乙酸(色谱纯)。超声波清洗器(EQ-500);DHG-9240A型电热恒温鼓风干燥箱(上海鸿都电子科技有限公司);BPZ-6090Lc型真空干燥箱(上海一恒科学仪器有限公司);高效液相色谱仪(安捷伦1260);色谱柱:安捷伦poroshell 120 EC-C18柱(4.6mm×50mm,2.7μm);质谱(API3200)美国AB公司。1.2 色谱条件色谱柱:以十八烷基硅烷健合硅胶为填充剂(柱长15cm,内径4.6mm,粒径2.7μm )或同等性能的色谱柱;柱温:40℃;流速:0.5 ml/min;进样量:5μL ;流动相:流动相A:称取0.386g乙酸铵,加水500mL溶解,用乙酸调pH至3.5;流动相B:乙腈,梯度洗脱:时间(分钟)流动相A(%)流动相B(%)0.0090100.00-5.0090→8010→205.00-9.0080→9020→101.3质谱条件1.3.1以电喷雾电离源(ESI)阳离子模式,气帘气20psi,碰撞气6,喷雾电压5500V,离子源温度600°C,雾化气是50 psi,辅助气是50 psi,时间9min。1.3.2 16种氨基酸的质谱参数,见表1。表1 16氨基酸的质谱参数氨基酸Q1Q3TIMEDPEPCECXP丙氨酸90.144.2353510153精氨酸175.2116353010153天冬氨酸134.474354110153胱氨酸241.174.11002610153谷氨酸148.2102353010153组氨酸156.1110353510163亮氨酸132.386.1352610113异亮氨酸132.286352610113赖氨酸147.3130353010153蛋氨酸150.1104352510103苯丙氨酸166.4120352510143脯氨酸116.270353510143苏氨酸120.174.3354110153酪氨酸182.3136353510153缬氨酸118.272354110153丝氨酸106.160.23530101331.4 标准品溶液的配制量取16种氨基酸的混合标准品1ml,置于50ml容量瓶,加0.1%甲酸水定容,摇匀即得标准品贮备液。1.5 供试品溶液的制备 精密称取样品约0.1g,置于25ml磨口的具塞比色管内,加6mol/L盐酸15ml,加入0.2g苯酚,用旋转混合仪和超声仪使样品充分分散并溶解,充氮气,盖紧塞子,置于110℃±1℃的恒温干燥箱内,水解22小时,取出冷却,过滤,用纯化水冲洗比色管,将水解液全部转移至50mL容量瓶中,用纯化水定容至刻度,摇匀,精密吸取1mL于10mL容量瓶中,置于真空干燥箱内,于40℃~50℃减压干燥(真空干燥箱内放入五氧化二磷作为干燥剂),干燥后残留物用0.1%甲酸水定容至刻度,摇匀,经0.45μm的微孔滤膜过滤,即得。1.6 线性实验将标准品贮备液稀释成0.004nmol/μl,0.008nmol/μl,0.012nmol/μl,0.016nmol/μl,0.020nmol/μl浓度梯度,每个浓度以5μL注入色谱系统。1.7 精密度实验将标准品贮备液稀释至一定的浓度,按色谱、质谱条件进行测定,连续进样6次。1.8 加样回收率实验精密称取五分的供试品,每份加入0.020nmol/μl标准品溶液5ml,定容。以5μL注入色谱系统。1.9供试品的测定 把1.5制备好的供试品溶液,以5μL注入色谱系统。以标准液的峰面积和浓度,通过外标法做曲线算出样品的浓度。2 结果与讨论2.1 色谱行为由于保健品中的氨基酸,用高效液相荧光法要进行柱前衍生,成本很高而采用液质联用法,不需要衍生,可大大节约成本,用流动相(0.35g乙酸铵加水0.5L溶解,然后加入0.5L乙腈,混合,用乙酸调pH=3.5)时,5min就能把16种氨基酸全部流出。采用液质联用法,通过MRM模式进行母离子及相应的子离子进行检测,能达到准确的分离和定量,见图1 。 从左到右,分别为丙氨酸、精氨酸、天冬氨酸、胱氨酸、谷氨酸、组氨酸、亮氨酸、异亮氨酸、赖氨酸、蛋氨酸、苯丙氨酸、脯氨酸、苏氨酸、酪氨酸、缬氨酸、丝氨酸。图1 16种氨基酸标准品的碎片离子色谱峰2.2 质谱行为在ESI(+)阳离子检测方式下,16种氨基酸的质谱定量分析,在质谱条件下,失去NH3形成+ 峰,或失去HCOOH重排生成+ 峰,从而进行色谱分离。2.3 线性回归方程和最低检测限以标准品的5个梯度浓度与其峰面积进行线性回归,拟合线性回归方程为Y=aX + b,相关系数r2 及最低检测限(S/N(信噪比)=3时,可测得标准品的最低检测限)见表2。表2 16种氨基酸线性回归方程、相关系数、最低检测限氨基酸Y=aX + b相关系数(r2)最低检测限(nmol/μl)丙氨酸Y=3.09e+006X + 6.95e+0030.99810.0013精氨酸Y=2.31e+007X + 1180.99990.0005天冬氨酸Y=3.45e+006X + 1.07e+0040.99760.0008胱氨酸Y=8.44e+005X + 1.72e+0030.99660.0006谷氨酸Y=7.6e+006X + 1.46e+0040.99840.0006组氨酸Y=7.58e+007X + 5.98e+0030.99990.0004亮氨酸Y=6.06e+007X + 5.24e+0040.99880.0001异亮氨酸Y=8.15e+007X + 6.26e+0040.99960.0001赖氨酸Y=1.24e+007X + 2.67e+0040.99780.0003蛋氨酸Y=1.34e+007X + 5.14e+0030.99890.0001苯丙氨酸Y=7.42e+007X + 4.08e+0040.99910.0001脯氨酸Y=7.9e+007X + 5.57e+0040.99970.0003苏氨酸Y=9.67e+006X + 2.49e+0040.99770.0003酪氨酸Y=1.76e+007X + 2.89e+0040.99920.0003缬氨酸Y=2.41e+007X + 3.31e+0040.99740.0003丝氨酸Y=1.06e+007X + 2.63e+0040.99810.00132.4 方法的精密度和重复性 取浓度0.012nmol/μl的16种氨基酸混合标准品连续进样6次。计算峰面积和保留时间的RSD值。结果见表3。表3 16种氨基酸峰面积和保留时间的相对标准偏差氨基酸峰面积RSD(%)保留时间RSD(%)丙氨酸4.480.57精氨酸4.670.34天冬氨酸4.570.44胱氨酸3.780.56谷氨酸3.360.24组氨酸3.330.13亮氨酸2.750.1异亮氨酸2.930.23赖氨酸4.790.43蛋氨酸3.920.09苯丙氨酸2.170.11脯氨酸4.540.28苏氨酸4.110.37酪氨酸3.610.13缬氨酸2.350.23丝氨酸3.280.162.5加样回收率实验,见表4。表4 16种氨基酸峰的回收率实验结果氨基酸平均回收率(%)RSD(%)丙氨酸94.114.05精氨酸82.912.73天冬氨酸82.694.66胱氨酸83.153.95谷氨酸98.234.1组氨酸78.671.61亮氨酸98.284.99异亮氨酸102.713.03赖氨酸98.584.49蛋氨酸95.983.34
不知哪位做过半胱氨酸盐酸盐的其他氨基酸。供试品溶液的制备 :取本品,加2%N-乙基顺丁烯二酰亚胺溶液制成每1ml中含4mg的溶液,作为供试品溶液;对照液的制备:另精密称取胱氨酸对照品10mg,加0.1mol/L盐酸溶液3ml,超声,加水适量使溶解,并加水使成100ml,摇匀,精密量取1ml,加2%N-乙基顺丁烯二酰亚胺溶液4ml,摇匀,作为对照品溶液。这里要问为什么对照液不直接拿供试品溶液稀释呢?而用胱氨酸对照品呢??不明白[em06] [em06]
[color=#444444]请教各位高手:调味品中的谷氨酸钠的含量检测一般都用甲醛法,现在我的样品是经过高温熬制的,里面的谷氨酸钠很可能变成了焦谷氨酸钠,这样用甲醛法检测的结果是否会受到影响?增高还是降低?[/color]
各位大侠好:有没有做过测试乳品中糠氨酸含量的检测实验的?不知道大家用的是什么方法?我现在也在测试糠氨酸的含量,但是不知道方法,请大家给予帮助!谢谢大家,如果有合适的资料可以发到我邮箱里 lhx0608@163.com谢谢大家,急盼大家的回复!
对照品进样量为20ul,供试品进样量为10ul,最后算含量的时候是否需要乘上供试品/对照品进样量的比值
我用高效液相色谱法测盐酸赖氨酸含量时,色谱柱:Agilent 氨基键合硅胶柱 250×4.6mm, 5μm,以0.05mol/L磷酸二氢钾溶液-乙腈溶液〔乙腈-水(10:1)〕(30:70)为流动相,检测波长为203nm,对照品不出峰,有经验的同学指导下啊!
请教大家,在sigma买的对照品有没有确定的含量啊,报告单上显示是大于等于99..0%,那在做含量测定用时对照品的含量按什么算啊?谢谢啊
国外对照品,说明书上说按无水物计算含量为XX,却没有说明水分多少,该如何处理?
关于液相色谱法检测食品中酪氨酸含量的具体方法有人知道吗?? 具体步骤是什么啊??
用于含量测定的对照品有无有效期?我看很多对照品标签上都没有标明,只有批号和含量
例如:头孢呋肟酯对照品,HPLC含量测定用时,含量96.9%,而用于UV检测溶出度时,含量为98.8%,为什么一个对照品有两个含量?检测方式不同么?
山葡萄酒含有22种氨基酸,其中人体必需的8种氨基酸(指人体自身不能合成的氨基酸)有苏氨酸、缬氨酸、异亮氨酸、亮氨酸、苯丙氨酸、赖氨酸、蛋氨酸、色氨酸等,总量为1350mg/L,占总氨基酸含量的45.6%,是一般葡萄酒的10~20倍;而一般葡萄酒只含有6~7种人体必需的氨基酸。
文/刘志军 华测检测[align=left]一泓清可沁诗脾,鲜爽茶感缘何来。优质的绿茶会有一种“鲜爽”的风味,喝茶的人对茶叶的鲜爽感应该不会陌生。那么,究竟是哪类成分物质让人有如此沁人心脾的感觉呢?[/align][align=left][color=black]不同品种茶叶泡出来的茶口感会有差异,例如绿茶的清香爽口、红茶的醇厚浓烈、黑茶的陈味香醇。导致不同品种的茶口味差异的主要原因是各类茶叶中的主要成分含量有所不同,[/color]茶叶的成分中主要包括有水、蛋白质、氨基酸、咖啡因、多元酚类、碳水化合物、脂质、矿物质、植物色素、维生素、挥发性成分、有机酸等。各成分含量多少与茶的口感息息相关,如:[color=black][/color]鲜味:主要成分为氨基酸,鲜中带甜,细嫩的茶叶中含量高。涩味:主要成分为多酚类物质。甜味:主要成分为可溶性糖及部分氨基酸。苦味:主要成分为咖啡碱、花青素、茶叶皂素。[/align][align=left] [/align][align=left][b]了解茶氨酸[/b][/align][align=left]上面我们讲到茶叶中各成分与茶口感的关系,原来茶的鲜爽感主要是因为茶叶中的氨基酸。氨基酸有很多种,绿茶中的鲜爽口感主要是一种叫“茶氨酸”的成分在发挥重要作用。茶氨酸是茶叶中特有的游离氨基酸,纯的茶氨酸为白色针状体,易溶于水,具有甜味和鲜爽味,是茶叶滋味的组分。茶氨酸在化学构造上与脑内活性物质谷酰胺、谷氨酸相似,是茶叶中生津润甜的主要成份。茶氨酸含量因茶的品种、部位而变动,干茶中茶氨酸含量约为新茶的1~2%左右,其含量随发酵过程减少,这就是为什么陈茶或者发酵茶的口感更偏醇厚浓烈,而绿茶的口感更清新鲜爽。[/align][align=left]1950 年,日本学者酒户弥二郎从绿茶中分离出了产生这种风味的主要物质——一种非蛋白质氨基酸,命名为茶氨酸。茶中的茶氨酸是左旋的,按照命名法记为“L-茶氨酸”。此后的研究发现,茶氨酸不仅为茶带来鲜爽风味,它本身还具有许多生理功能。比如它能突破血脑屏障直接影响大脑活动,从而对人的情绪产生影响。有研究表明茶氨酸对大脑各部位单胺类代谢影响时发现,茶氨酸可以促进脑中枢多巴胺释放,提高脑内多巴胺生理活性。多巴胺是一种活化脑神经细胞的中枢神经递质,其生理活性与人的情感状态密切相关。尽管人们对茶氨酸在大脑中枢神经系统的作用机制并不是十分清楚,但茶氨酸对精神和情感的影响无疑有部分是来自于对中枢神经递质多巴胺生理活性的作用,当然饮茶抗疲劳作用也被认为在一定程度上可能来自这一效果。[/align][align=left][/align][align=left][b]什么样的茶富含茶氨酸?[/b][/align][align=left]作为饮料,“好茶”的根本标准还得是“好喝”,而茶氨酸以及游离氨基酸的含量与茶的风味呈正相关,也就是说茶氨酸和游离氨基酸含量高的茶,往往会更好喝。[/align][align=left]茶氨酸的合成跟茶树的光合作用、生长环境温度密切相关。如果光照不足、或者温度较低,那么茶氨酸的分解就会受到抑制,茶的芽和叶中就会积累比较多茶氨酸。春天茶叶发育期间温度较低,所以茶氨酸的分解以及儿茶素的合成受到抑制。这样春茶中茶氨酸含量高而茶多酚含量相对低。而夏茶和秋茶生长采摘时温度较高,光合作用旺盛,相对而言茶氨酸含量低而茶多酚含量高,口感也就与春茶不同。[/align][align=left]茶叶中的茶氨酸含量一般为1%~4%左右,富含茶氨酸的茶叶主要有三类。[/align][align=left]第一类来自于品种优势,例如安吉白茶和福鼎白茶,都有明显的品种优势。安吉白茶的游离氨基酸含量测定显示最高可达8%,说明其富含茶氨酸。[/align][align=left]第二类来自于环境优势,比如高山茶中的庐山云雾茶、阿里山乌龙茶、杉林溪乌龙茶、奇莱山乌龙茶都具有“高茶氨酸”的特征。高山茶在生长的过程中经常处于云雾中,阳光受到较多遮挡,且高海拔区域的气温低于低海拔的区域,这类茶中的茶氨酸含量也就相对较高。[/align][align=left]第三类来自于工艺优势,日本人常用遮荫的方法来提高茶叶中茶氨酸的含量,以增进茶叶的鲜爽味。日本抹茶在生产时采用人工遮阴的方法,目的就是减弱茶叶生长过程光合作用而使得茶氨酸含量大大提升。当年酒户弥二郎分离茶氨酸,用的玉露茶就是通过人工干预茶叶生长过程中的光合作用而得来的。[/align][align=center][b] [/b][/align][align=left][b]怎样才能得到可以作为“食品原料”的茶氨酸呢?[/b][/align][align=left]不管是作为食品添加剂、膳食补充剂,还是食品原料,都需要相对纯度较高的茶氨酸。中国批准作为新食品原料的茶氨酸对纯度的要求为≥20%。这大大高于茶中茶氨酸的含量——也就是说,把茶氨酸分离提纯,是它实现这些用途的前提。目前,可采用的分离方法有:沉淀法、离子交换树脂法、膜分离法、化学合成法、生物发酵法、酶转化法以及植物细胞培养法等。[/align][align=left]前面说到茶氨酸易溶于水,那是不是只需要用水浸泡就可以提取出来呢?事情并没那么简单。茶氨酸溶于水的同时,茶多酚、咖啡因等各种其它成分同样会溶于水中。要分离出高纯度的茶氨酸,还需要除去其它我们不想要的成分。目前,工业上可采用的分离方法有三种:沉淀法、离子交换树脂法和膜分离法。[/align][align=left]沉淀法:是最传统的提取方法,就是通过改变提取时的温度、酸碱环境把混合物中的一种或几种成分充分地沉淀下来再进行过滤去除,从而把我们期望留下的茶氨酸和其他成分进行分离。这种手段优点是操作简单,缺点是流程比较长,步骤较多,在提取的过程中容易引人其他的有害物质。[/align][align=left]离子交换树脂法:大致原理是通过离子交换树脂,把茶氨酸吸附到树脂上,而让其他成分流过。再用溶液把茶氨酸从树脂上“洗脱”下来,就得到了茶氨酸含量大大提高的“粗品”。当然这类粗品可能含有的有害物质残留还比较多,所以需要再纯化之后得到高含量和安全性兼顾的茶氨酸成品。此方法的优点是提取的茶氨酸纯度高,缺点是成本相对传统提纯方法高。[/align][align=left]膜分离法:是现代天然产物分离中的新兴技术,在生产加工饮用水领域应用广泛,原理就是利用通过半透膜孔径把不想要的有害物质过滤掉。选择合适的过滤膜孔径是关键,可以把分子比茶氨酸大的和小的成分都去除,而只留下茶氨酸和分子大小与它接近的成分。膜分离法的优势在于不引入其他的物质,劣势是与目标分子大小相当的物质很难去除,单纯利用此方法提纯也很难获得高纯度的产品,而且此方法目前很难实现量产提纯茶氨酸。[/align][align=left]除此之外,生产茶氨酸还有化学合成法、微生物发酵法、细胞培养法,但是这些方法都各有其优势和局限性。[/align][align=left]茶叶中的茶氨酸含量本来就不高,受技术限制,目前要量产茶氨酸就会面临提取成本高的问题,再加上如果从茶叶中只是提取茶氨酸的话,经济效益就很低。春季茶树鲜叶茶多酚含量较高,做出的优质春茶往往能卖出更好的价格,也就不会用来提取茶氨酸了。不过,茶中有经济价值的成分并不止茶氨酸,比如茶多酚、咖啡因含量高,也更有提取价值。可以在提取茶多酚时产生的废液中合理利用其中含有的茶氨酸,作为一举多得的附加值产品,由此得到的茶氨酸的成本自然就跟着降低了。[/align][align=left][b][/b][/align][align=left][b]茶氨酸的安全性及法规管控[/b][/align][align=left]有科研人员对茶氨酸进行安全性实验,结果表明茶氨酸的大鼠急性毒性在5g/kg以上。他们对大鼠每天服用2g/kg茶氨酸在连续28天的亚急性毒性实验中没有观察到任何毒性反应。此外,在突然变异的实验中也没有发现茶氨酸的任何诱变作用。[/align][align=left]在日本,对茶氨酸的摄入量没有限制。1964年,日本批准了L-茶氨酸作为食品添加剂使用。而美国FDA也在1985年给予了L-茶氨酸GRAS的分类,意味着可以根据需要用于各种食品中。中国在2014年7月18日,原国家卫计委批准了它作为新食品原料。原国家卫计委关于批准茶叶茶氨酸作为新食品原料的公告(2014年第15号)做了如下规定:[/align][align=left](1)必须以茶叶为原料,经提取、过滤、浓缩等工艺制成;----这意味着化学合成法和生物发酵法制成的茶氨酸不能用作新食品原料;[/align][align=left](2)每日食用量≤0.4 克/天;[/align][align=left](3)对产品质量的要求,性状为黄色粉末、茶氨酸含量≥20g/100g、水分≤8g/100g,也就是作为新食品原料的茶氨酸含量(纯度)必须≥20g/100g;[/align][align=left](4)使用范围不包括婴幼儿食品,卫生安全指标应符合国家相关标准规定。[/align][align=left]我国现行有效的茶氨酸产品标准《QB/T 4263-2011 L-茶氨酸》中对采用生物发酵法、酶法转化或提取精制而得的L-茶氨酸的感官、理化、含量、重金属、微生物等十几个指标制定了限量要求。茶氨酸相关指标的检测方法有《QB/T 4263-2011 L-茶氨酸》中的附录B和《GB/T 23193-2017 茶叶中茶氨酸的测定高效液相色谱法》。[/align][align=center][color=black] [/color][/align][align=left][b]茶氨酸在食品加工中的应用[/b][/align][align=left]茶氨酸在日本、美国可作为食品添加剂,目前在我国可用作新食品原料使用(需要注意每日食用量和适宜人群)。茶氨酸具有优良的加工特性和稳定性,可以广泛应用于各类点心、糖果及果冻、饮料、口香糖等食品中。总的来说,茶氨酸主要应用在如下领域:[/align][align=left]1)作为茶饮料的品质改良剂,在茶饮料生产过程中添加一定量的茶氨酸,能明显改善茶饮料的品质和风味;[/align][align=left]2)作为改善食品风味的原料,研究表明,茶氨酸可改善咖啡、可可、果蔬饮料、啤酒等的苦味,减轻葡萄酒的涩味,因此,可作为这些食品的风味改良剂。[/align][align=left]茶氨酸在常规的食品加工条件下(如杀菌、pH和加热等)比较稳定,应用范围广。目前日本已开发出添加茶氨酸的巧克力、果冻、布丁、口香糖、保健茶和各种清凉饮料。茶氨酸功能作用这么好,但在食品生产加工过程中同样要注意茶氨酸原料是否达到食品级要求以及其他安全指标是否达标国家管控要求。[/align]
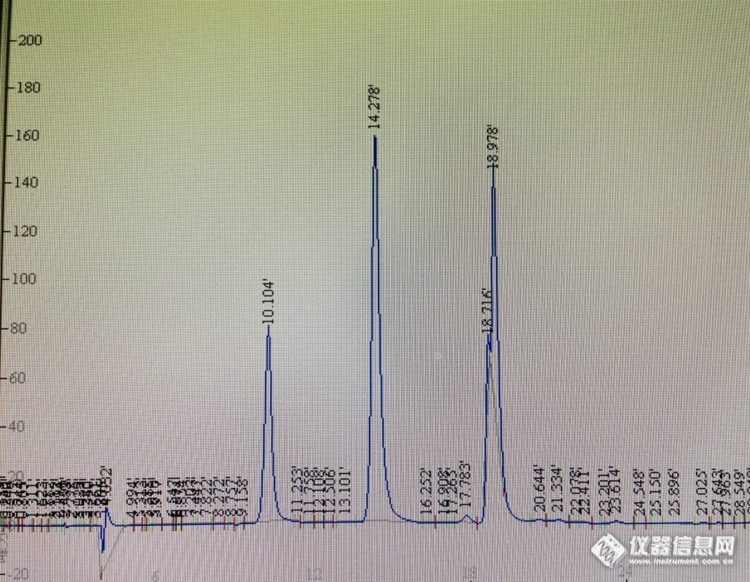
柱子是迪马钻石 C18 5um 250*4.6mm 柱体积4.2ml 流动相和对照品的制备全部按照药典规定进行:以乙腈-0.1mol/L醋酸钠溶液(用醋酸调节pH值至6.5)(7:93)为流动相A,以乙腈-水(4:1)为流动相B,按下表中的规定进行梯度洗脱;检测波长为254nm;柱温为43℃。理论板数按L-羟脯氨酸峰计算应不低于4000。时间(分钟)流动相A(%)流动相B(%)0~1111~13.913.9~1414~2929~30100→9393→8888→8585→6666→00→77→1212→1515→3434→100对照品溶液的制备取L-羟脯氨酸对照品、甘氨酸对照品、丙氨酸对照品、L-脯氨酸对照品适量,精密称定,加0.1mol/L盐酸溶液制成每1ml分别含L-羟脯氨酸80μg、甘氨酸0.16mg、丙氨酸70μg、L-脯氨酸0.12mg的混合溶液,即得精密量取上述对照品溶液和供试品溶液各5ml,分别置25ml量瓶中,各加0.1mol/L异硫氰酸苯酯(PITC)的乙腈溶液2.5ml,1mol/L三乙胺的乙腈溶液2.5ml,摇匀,室温放置1小时后,加50%乙腈至刻度。摇匀。取10ml,加正己烷10ml,振摇,放置10分钟,取下层溶液,滤过,取续滤液,即得。[
我们一般做含量时,是取两份对照品,一份进5针,一份进2针,第一份5个数据计算RSD值, 而我们现在要进行技术比武,它的标准是两份对照各进3针,这能计算RSD值吗?还是计算其他数据?你们遇到过吗?[em0812][em0812]
有谁跑过盐酸组氨酸的薄板,对照溶液为自身样品溶液(50mg/ml)稀释而成(0.1mg/ml),以正丁醇-丙酮-浓氨溶液-水(10:10:5:2)为展开剂。显色后发现对照的斑点位置高于供试品。按说自身对照的话,Rf值应该是一样的啊,请问各位大虾,这是怎么回事啊?[em53] [em53]
中国药典中,某品种项下规定含量测定需使用某对照品,但是中检所仅提供鉴别用的,没有含量测定用的对照品。可以直接用这个鉴别对照品去测含量吗?
请问哪位前辈有下表中的乳与乳制品中动物水解蛋白鉴定-L(-)-羟脯氨酸含量测定(检测方法由中国检验检疫科学院食品安全所提供。这个文本啊!附件1食品中可能违法添加的乳与乳制品中动物水解蛋白鉴定-L(-)-羟脯氨酸含量测定(检测方法由中国检验检疫科学院食品安全所提供。非食用物质名单(第二批) 序号名称主要成分可能添加的主要食品类别可能的主要作用检测方法1皮革水解物皮革水解蛋白乳与乳制品含乳饮料增加蛋白质含量乳与乳制品中动物水解蛋白鉴定-L(-)-羟脯氨酸含量测定(检测方法由中国检验检疫科学院食品安全所提供。联系方式: Wkzhong@21cn.com)2溴酸钾溴酸钾小麦粉增筋GB/T 20188-2006 小麦粉中溴酸盐的测定 离子色谱法3β-内酰胺酶(金玉兰酶制剂)β-内酰胺酶乳与乳制品掩蔽抗生素液相色谱法(检测方法由中国检验检疫科学院食品安全所提供。联系方式: Wkzhong@21cn.com)4富马酸二甲酯富马酸二甲酯糕点防腐防虫气相色谱法(检测方法由中国疾病预防控制中心营养与食品安全所提供)







 我要推广仪器
我要推广仪器
 下载APP
下载APP