我现在要用HPLC测定亚甲基双水杨酸中微量的水杨酸含量,可是两种物质性质几乎是相同的,在样品处理很难。请大家帮我出谋划策一下,用什么方法能萃取亚甲基双水杨酸中微量的水杨酸?
摘要:甲基汞(MeHg)是1种毒性很强的物质,可以通过水生食物链累积和富集,并最终危害处于食物链顶端的人类。基于甲基汞暴露对人体健康危害的研究,文章介绍了几个甲基汞暴露风险评价指标(甲基汞参考剂量、甲基汞临时性周可承受摄入量、职业汞暴露评价指标)以及发达国家在预防甲基汞暴露(主要为食用鱼类)方面的经验。1引言甲基汞是1种具有较强神经毒性的污染物质。20世纪}0年代发生在日本的水误病事件,使人们首次认识到甲基汞的毒性会对人体健康造成严重影响。类似的环境污染公害事件(食用了甲基汞含量超标的水产品或谷物)也在美国、伊拉克、巴西、印度尼西亚以及我国松花江流域等地发生。本文以甲基汞暴露与人体健康影响之间的关系为着眼点,分析了甲基汞暴露对人体神经系统、心血管系统和免疫系统的毒害影响,详细介绍了发达国家在甲基汞健康风险评价指标的设定和鱼类体内甲基汞含量标准制定方面的研究进展。......[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=104692]甲基汞暴露与人体健康影响[/url]
摘要:甲基汞(MeHg)是1种毒性很强的物质,可以通过水生食物链累积和富集,并最终危害处于食物链顶端的人类。基于甲基汞暴露对人体健康危害的研究,文章介绍了几个甲基汞暴露风险评价指标(甲基汞参考剂量、甲基汞临时性周可承受摄入量、职业汞暴露评价指标)以及发达国家在预防甲基汞暴露(主要为食用鱼类)方面的经验。关键词:甲基汞 暴露 风险评价 鱼类食用警示1引言甲基汞是1种具有较强神经毒性的污染物质。20世纪}0年代发生在日本的水误病事件,使人们首次认识到甲基汞的毒性会对人体健康造成严重影响。类似的环境污染公害事件(食用了甲基汞含量超标的水产品或谷物)也在美国、伊拉克、巴西、印度尼西亚以及我国松花江流域等地发生。本文以甲基汞暴露与人体健康影响之间的关系为着眼点,分析了甲基汞暴露对人体神经系统、心血管系统和免疫系统的毒害影响,详细介绍了发达国家在甲基汞健康风险评价指标的设定和鱼类体内甲基汞含量标准制定方面的研究进展。2甲基汞暴露汞的毒性取决于化学结构,汞暴露有3种形态:元素汞、无机汞和有机汞(以甲基汞为主),它们的中毒症状和暴露途径各不相同。饮食(特别是鱼类)是甲基汞暴露的主要途径[1];医用汞齐和吸人性暴露则是元素汞蒸汽暴露的主要途径,环境中不同途径的汞暴露量[2]见表1。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=99627]甲基汞暴露与人体健康影响[/url]
硫化氢亚甲基蓝监测时标曲斜率很低,显色液的问题还是标准溶液的浓度问题,我们是买的硫化氢标液,但是是以前没用完的。最高的吸光度才0.263线性很好就是斜率无语了
现测量水中二甲基亚砜的含量,采用的方法是将含微量二甲基亚砜的水加入到高锰酸钾(25ml)和硫酸(5ml)的混合溶液中,滴上指示剂,然后用硫代硫酸钠来滴定。现在的问题是如果含微量亚砜的水中有少许的氨,用这样的方法滴定,是不是存在偏差,使得测试的结果,亚砜含量偏高呢,请高手指教。
之前在论坛上看见有人在说亚甲基蓝分光光度法大二斜率一般是在0.018-0.020之前,我做出来的斜率是0.011,截距为0.000,R=0.999,差别很大啊,是什么原因啊,这样的曲线能用吗?急求解答
在gb/t 12496.10-1999中的操作步骤:“称取经粉碎至71 dam的干燥试样0. 100 g(称准至1 mg),置于100 mL具磨口塞的锥形烧瓶中,用滴定管加入适量的亚甲基蓝试验液,待试样全部湿润后,立即置于电动振荡机上振荡20 min,环境温度(25士5)'C,用直径12.5 cm的中速定性过滤纸进行过滤。将滤液置于光径为1 cm的比色皿中,用分光光度计在波长665 nm下测定吸光度,与硫酸铜标准滤色液〔称取4. 000 g结晶硫酸铜溶于1 000 m1,水中〕的吸光度相对照,所耗用的亚甲基蓝试验液的毫升数即为试样的亚甲基蓝吸附值。”我初次用分光光度计,不知道怎么对照法?请高人指点!~~~
版友问题:请问二甲基亚芳基硅氧烷为固定液的色谱柱是?气相色谱柱。
[color=#444444]已知用5%己内酰胺-四氢化萘为原料,加氢还原制备六亚甲基亚胺(七元杂环亚胺)沸点138℃,通过用填充柱[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析发现溶剂四氢化萘平头,如果想定量分析反应后产物六亚甲基亚胺的量是用外标法还是内标法?内标物或外标物该选什么呢?[/color]
有没有人做过水产品中N-二甲基亚硝胺的实验,求高效方法?国标5009.26太难操作了,要取200g肉蒸馏萃取浓缩,才做得出来,而且这个物质容易分解,回收率会很低,求各位大神提供意见!
检测活性碳,用亚甲基蓝检测吸附值时,硫酸铜(所有按标准配制)的吸光度一般为多少?我只有0.08,是不是太低了?亚甲基蓝标定浓度后,按浓度计算吗?(用浓度代替1.5计算?),要注意哪些?我的数据是60-80mg/g,供货报告单为160,相差太远了,不知道是什么原因?
亚甲基蓝分光光度法测阴离子表面活性剂的时候 萃取完之后 整个瓶的里上下两层都变无色了 亚甲基蓝褪色了 啥原因 样品取小了 还是一样
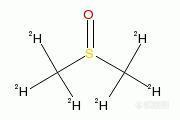
[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911082356_182710_1610969_3.jpg[/img]二甲基亚砜 基本信息 英文名 Dimethyl sulfoxide 别名 Dimethylsulfoxide Methyl sulfoxide Sulfinylbis (methane) DMSO 产品名称 二甲基亚砜 分子式 C2H6OS 分子量 78.12 CAS 登录号 67-68-5 EINECS 登录号 200-664-3 FEMA 登录号 3875 物理化学性质 密度 1.1 熔点 18.4-19.0°C 沸点 189°C 折射率 1.477-1.48 闪点 95°C ⒈概述 二甲基亚砜(DMSO)是一种含硫有机化合物,分子式为(CH3)2SO,常温下为无色无臭的透明液体,具有吸湿性的可燃液体,既有高极性,高沸点,非质子,于水混溶的特性,毒性极低,热稳定性好,能溶于乙醇,丙醇,苯和氯仿等大多、数有机物,被誉为“万能溶剂”。 二甲基亚砜广泛用作溶剂和反应试剂,特别是丙烯腈聚合反应中作加工溶剂和抽丝溶剂,作聚氨酯合成及抽丝溶剂,作聚酰胺,聚酰亚胺和聚砜树脂的合成溶剂,以及芳烃,丁二烯抽提溶剂和合成氯氟苯胺的溶剂等。除此之外,在医药工业中二甲基亚砜还有直接用作某些药物的原料及载体。二甲基亚砜本身有消炎止痛,利尿,镇静等作用,亦誉为“万灵药”,常作为止痛药物的活性组分添加于药物之中。
水质 阴离子表面活性剂的测定 亚甲蓝分光光度法 疑问求解 空白的氯仿萃取后有明显蓝色,650nm吸光度值达0.23,而且阴离子表面活性剂浓度和吸光度值无线性关系?亚甲基蓝在氯仿中会溶解显现蓝色吗?亚甲基蓝配制前需要用氯仿萃取多长纯化吗?麻烦各位前辈指点!
最近测试聚硅氧烷中六亚甲基二异氰酸酯(HDI)含量,简单总结就是不同的进样口温度导致样品中HDI峰响应不同。参考GB/T18446,溶剂均为乙酸乙酯,用邻苯二甲酸二乙酯做内标和HDI配标液测相对校正因子,取1g样品和邻苯二乙酯测HDI含量。用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]测,根据标准进样口温度设为125℃时,标液中HDI(3mg/ml)峰面积为900-1000,样品中HDI峰面积为6-8,含量约0.03%。若进样口温度为250℃,标液中HDI(3mg/ml)峰面积也差不多在900-1000,但是样品中HDI峰面积增至200-300,含量约1%.用质谱测,不同进样口温度同样呈现该趋势,且用质谱识别,小面积的峰(125度进样口,峰面积6-8那个)和大面积的峰(250度,峰面积900-1000那个)都是六亚甲基二异氰酸酯。所以为什么进样口温度对标液中的HDI峰面积影响不大但是对样品中HDI峰面积影响这么大呢?哪个是准确的呢?高温发生聚合?但为什么250度时大面积的峰也能识别出是HDI呢?期待得到大家的指点!
新手求助,GB 5009.26N-二甲基亚硝胺的测定中,最后附的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]图,标出了两个峰,NDEA和NDMA,NDMA是N-2甲基亚硝胺,NDEA不是N-二乙基亚硝胺吗?最后计算结果要取这两个峰之和吗?还是NDEA不作计算?
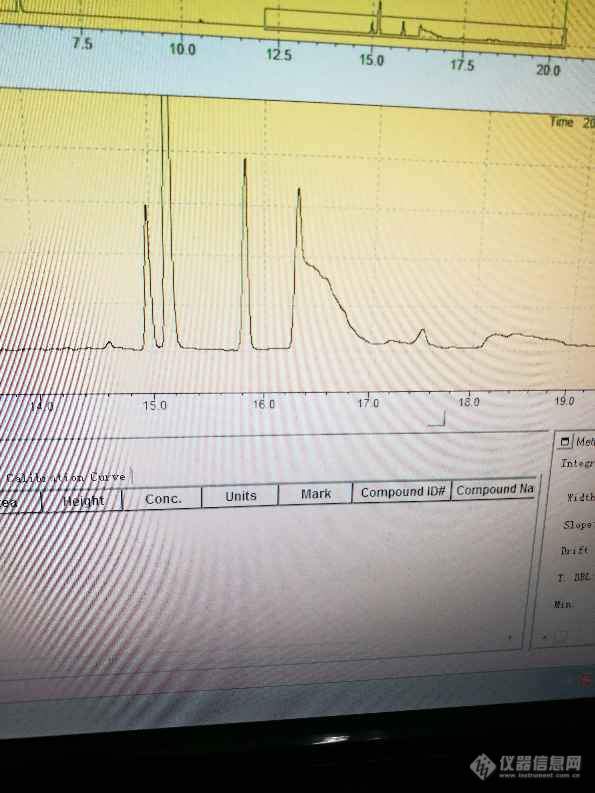
[color=#444444]本人用N甲基吡咯烷酮做稀释溶剂,检测二甲基亚砜的溶残,发现二甲基亚砜的峰形很差,拖尾严重,空白溶液中二甲基亚砜这个位置是没有峰的,换了个色谱柱,仍然出现二甲基亚砜峰形差,拖尾严重的现象,请问是什么原因造成的呢,二甲基亚砜的谱图是第四个峰[/color][color=#444444][img=,595,793]https://ng1.17img.cn/bbsfiles/images/2019/06/201906241634207260_5339_1827556_3.jpg!w595x793.jpg[/img][/color]
亚甲基蓝法测阴离子表面活性剂,石英皿装好氯仿萃取液,放入仪器,不去动它,可吸光度总是不停往下跳,无法稳定下来。仪器是新买的国产普析,才5万块。不知是仪器问题,还是说亚甲基蓝络合物在不断分解。
如文件为亚甲基蓝的红外标准谱图,小弟粗略地分析了下,还请大虾指点,看是否有问题?
有没有大虾做过肉中的N-二甲基亚硝胺的实验,求高效方法?国标中的方法(气质法)好变态,不太适应大批量的操作(一个个蒸馏,然后液液萃取三次,浓缩),灵敏度太低,要取200g肉才做得出来,求各位高手提供意见!
有时叠氮化物的亚甲基会分裂成两个一样的四重峰(化学位移略有不同)而甲基会分裂成两个一样的三重峰,为什么
各位大神,我现在在做活性炭的亚甲基蓝测试,用的是木质活性炭的国标法,但是一直测不出结果。 第一个问题是控制不好亚甲基蓝的添加量;第二个问题是由于我的活性炭漂浮率太高,亚甲基蓝溶液滴进去就分层了,活性炭都漂浮在溶液上层,一直不湿润; 第三个问题是滤出来的溶液测分光度也测不出来,浓度太高了。第四个问题是,我购买的亚甲蓝是分析纯,那么纯度可以按99%计算吗?我按照99%的纯度计算出来,称量了与1.5g干燥亚甲基蓝相应的固体,60℃下搅拌半个小时溶解,也过滤了,但是测出的吸光度为0.122,与硫酸铜标准液0.379差了很多,是为什么呢?是没有完全溶解吗?还是说纯度不够,质量少了? 望回复,感谢
在醋酸介质中,求助亚甲基蓝与碘的反应方程式。(注:碘过量)
次甲基蓝和亚甲基蓝是不是一种东西?问了几个人答案不一致,大家讨论一下!听说过次氯酸 亚氯酸,应该是有区别的!大家的意见呢?
亚甲基蓝染色液是怎么配制的?若测COD在配制是需不需要加入其他的试剂?
3、 取血蓝蛋白(lemocyanin) 25mg, 溶于10mmol/L PBS (pH8.0)液中(III液) 4、 将II液与III液混合,在磁力搅拌下逐滴加入I液(余下0.5ml) 5、 室温下避光搅拌1小时,逐滴加入余下的I液 6、 4度搅拌12小时 ;7、 静置10小时(4度) 8、 有蒸馏水使之充分透析(约48小时),得免疫原。 3、孕酮与与β-半乳糖苷酶偶联的N-羟琥珀酰亚胺酯法 1、 用二垩烷(dioxane)溶解孕酮-11-半琥珀酸酯,配成浓度为100m mol/L的溶液。 2、 加羟琥珀酰亚胺(N-hydroxysuccinimide) 100 m mol/L 和 DCC(二环已基碳化二亚胺),200 m mol/L, 4度反应16小时。 3、 用簿层扫描方法纯化(氯仿:水=9:1) 4、 按孕酮/酶摩尔浓度比约为10的比例,将上述溶液加入到酶液(用pH7.4,浓度50 m mol/L的磷酸缓冲液溶解)中。 5、 (二)含有氨基或可还原硝基半抗原的偶联 1、芳香胺类半抗原与蛋白质重氮化偶联的操作步骤 6g1、 用0.1 mol/L HCl溶液配制 4 m mol/L浓度的半抗原。 2、 滴加1%NaNO2(过量),4度持续搅拌。NaNO2的加入量可用淀粉-碘化物试纸或在白色磁砖上加1%淀粉和50m mol/L KI进行监控。游离亚硝酸可将氧化物氧化成碘,碘再与淀粉反应变成蓝黑色。 3、 溶液变成蓝黑色后,继续反应15分钟。4、 用pH9.0、浓度为200m mol/L的硼酸或碳酸缓冲液溶解蛋白。 5、 边搅拌,边加入重氮化的半抗原(防止局部发生酸过量现象),调节pH到9.5。 6、 冰箱中搅拌反应2小时,不断调节pH到9.0。 7、 用PBS透析2天 8、 -20度保存(浓度为20mg/mL) 双功能的酰亚胺酯(imidate esters)可以氨基反应,形成脒。例如:用二甲基已二酰亚胺酯(dimethyladipimide)将去甲基三正喋呤(desmethylmortriptyline)与β-半乳糖苷酶偶联。2、、应用双功能酰亚胺酯(imidate esters)制备去甲基三正喋呤-与β-半乳糖苷酶标记特的操作步骤1、 用含5%(W/V)N-乙基吗啉的无水甲醇0.4ml,在室温下溶解570ug去甲基三正喋呤和488ug 二甲基已二酰亚胺酯(dimethyladipimide)(A液) 2、 取与β-半乳糖苷酶 100 ug, 溶于pH9.9的100 m mol/L碳酸缓冲液(含MgCl2 10m mol/L,2-巯基乙醇 10 m mol/L 0.1ml(B液)3、 将A液倒入B液。4、 20度反应90分钟后,加含NaCl 100 m mol/L, MgCl2 10 m mol/L和2-巯基乙醇 10 m mol/L、pH7.5的Tris-醋酸缓冲液(50m mol/L) 1ml, 终止反应。 5、 过sephadex G-25, 去除小分子物质,得酶标记物(约75%的酶与半抗原结合,但用三正喋呤代替去甲三正喋呤(demethylmortriptyline )进行偶联,则只有15%的酶与之结合。 (三)含巯基半抗原的偶联 可用马来酰亚胺方法与蛋白偶联。此外,将载体蛋白用溴乙酰胺(bromoacetamide)激活。或将载体蛋白与半抗原在pH4.0的醋酸缓冲液中,通过过氧化氢的作用形成二硫键,也可以将半抗原连接到蛋白质分子上。 (四)含羟基的半抗原偶联 醇类羟基通过形成半琥珀酸酯转化为羧基的操作步骤 1、 15g 2,2,2-三氯乙醇(2,2,2-trichloroethanol),12g 琥珀酸酐(succinic anhydride)和8.7ml 三乙基胺(triethylamime)用100 ml乙酰乙酯溶解。 2、 加热回流1小时。 3、 减压蒸馏去溶剂,,残余物用5% NaHCO3水溶液溶解。4、 用乙醚洗涤两次,然后用H2SO4进行s酸化(pH到2.0). 5、 用水洗涤固形物(为三氯乙基半琥珀酸酯)两次,用氯仿-已烷使其结晶(产量约75%,熔点88-89度) 6、 取2.5g 半琥珀酸酯溶于6.5ml 亚硫酰氯(thionyl chloride)中,65度加热30分钟。 7、 减压蒸发,干燥1小时(高度真空条件下)。 8、 将上述产生(2,2,2-三氯忆基琥珀酰氯)溶于15ml N,N-二甲基-乙酸乙酰胺(N,N-dimethylethylacetamide)中,室温搅拌反应2小时。 9、 65度真空蒸发后,用异丙醇使结晶析出来(得盐酸化的结晶---5`-酯约84%,熔点160度)。 10、用溶于二甲基甲酰胺中的锌和醋酸解离三氯乙酯,得f半抗原-半琥珀酸酯,这样引和的羧基可与蛋白质偶联(如用碳化二亚胺化)。 半抗原用NaIO4氧化其中的糖苷醇后再与蛋白质偶联的操作步骤 1、 20mg 腺苷溶于1ml 100m mol/L NaIO4溶液中,4度避光反应30分钟。2、 加1滴乙二醇(得A液) 3、 将A液加入到β-半乳糖苷酶液(20mg/ml,用150m mol/L NaCl,10m mol/L MgCl2水溶液溶解,用3%K2CO3调节pH至9.0)中 4、 4度反应2小时,期间不断调节pH9.0 5、 加入临时配制的50 mg/ml NaBO4溶液,用量为反应体积的1/10。 4度反应过夜。 6、 用含有MgCl2 10m mol/L,2-巯基乙醇 10 m mol/L、 NaCl 100 m mol/L的50 m mol/L磷酸缓冲液(pH7.4)透析(更换透析液数次) (五)含酮基或酮基半抗原的偶联是将酮基经羟胺类化合物处理变成肟类化合物,再进一步将肟类化合物中的羟基,衍变成羧基化合物,再进一步进行含羧基半抗原的偶联操作。这类羟胺类化合物主要有:氨氧乙酸aminoxy acetic acid 或羧甲氧胺carboxymethoxyl amine 或者盐酸羟胺酮基的类固醇分子中引入羧基的操作步骤 1、 在200ml 乙醇中,加入O-(羧甲基)羟胺 (O-(carboxyl)hydroxylamine)和酮基半抗原,使其浓度分别为10m mol/L 和4m mol/L H 2、 加热回流90分钟 3、 旋转蒸发,减少容积,然后加水至40ml,用乙醚抽提 4、 用水洗涤乙醚抽提物,用Na2SO4干燥成白色粉末。(六)、其他半抗原的偶联 虽含有游离基团,但因这些基团对于维其生物活性十分重要,因些不能直接用来与载体蛋白 偶联8制备雌二醇-6-肟的操作步骤 a、 雌二醇二醋酸盐的制备 1、 1g雌二醇溶于14ml 吡啶及3.5ml 醋酐中2、 加热回流1小时,冷却后倾入冰水中。 3、 收集白色晶体,得产物约1.1g(熔点126到127度)b、 雌二醇-6-酮-二醋酸盐的制备 4、雌二醇二醋酸盐1.1g,滴加冰醋酸3.8ml 溶解后加含0.93g CrO3的含水冰醋酸6.35ml (H2O:Hac=0.75:5.6) 5、室温搅拌1小时,静置24小时 6、用水稀释,用乙醚提取4次 7、用蒸馏水洗2次8、减压蒸馏,得结晶油状渣物。 9、用90度烘干20分钟,得粗制品约500mg 10、用11ml 无水乙醇溶解粗制品,再加1.1ml 冰醋酸及1.5g吉纳你特T试剂(Girad T),回流1小时。 11、冷却后,用冰致冷的蒸馏水稀释,用2.5mol/LNaOH调节pH至6.0-6.2. 12、用乙醚抽提3次,弃去乙醚。 13、水层用浓盐酸酸化15、用乙醚抽提3次。16、合并乙醚抽提液,用0.125mol/L碳酸钠溶液洗1次,用蒸馏水洗3次。
急用亚甲基兰脱色方法请教亚甲基兰脱色方法的详细步骤,感激不尽,谢谢!!!
测定乙酸乙酯残留,溶剂为二甲基亚砜,测定精密度,发现有一些溶剂峰没有出现,而有的溶剂峰却出现了。请教大家是怎么回事,应该主溶剂的峰都是应该出现的啊,而且很大。溶剂峰和乙酸乙酯峰分离度很好。乙酸乙酯出峰在10分钟,二甲基亚砜出峰在17分钟。
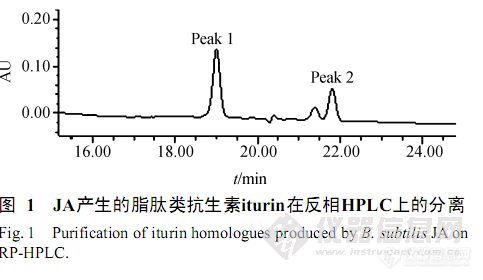
【作者】 陈华; 袁成凌; 蔡克周; 郑之明; 余增亮;【Author】 Hua Chen,Chengling Yuan,Kezhou Cai,Zhiming Zheng,Zengliang Yu(Key Laboratory of Ion Beam Engineering,Chinese Academy of Sciences,Hefei 230031,China)【机构】 中国科学院离子束生物工程学重点实验室; 中国科学院离子束生物工程学重点实验室 合肥230031; 合肥230031;【摘要】 枯草芽孢杆菌JA产生的抗生素对植物病原真菌具有广谱抗性,明确抗生素的种类是进一步研究的基础。用6mol/L盐酸沉淀JA菌株的去菌体培养基,再用甲醇抽提获得抗生素的粗提物。利用反相HPLC系统,将粗提物过DiamonsilC18柱,收集有抗小麦赤霉病等病原真菌活性的化合物1、2。运用电喷雾质谱法(ESI/MS)测得其分子量分别为1042.4D和1056.5D。再利用碰撞诱导解离(CID)技术获得化合物的典型结构特征离子碎片,结果表明分子量为1042.4D的化合物一级结构为Pro-Asn-Tyr-βAA-Asn-Tyr-Asn-Gln(βAA为14个碳原子的氨基脂肪酸),属于脂iturin A。化合物1、2为相差一个亚甲基(-CH2)的iturinA同系物。研究结果提供了一种从枯草芽孢杆菌发酵液中快速分离纯化和鉴定脂肽类抗生素iturin A的新方法。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301640_380623_2379123_3.jpg
我公司正在调试二甲基亚砜脱水精馏塔,现需一些关于这方面的资料。请有这方面经验的达人给予指导。帮忙介绍一下你们的工艺参数的设置以及时如何操作。我在google上收到如下的文献。请高手帮忙找一下关于二甲基亚砜精馏的文献(如下所示),不胜感谢。《应用间歇真空精馏技术回收高纯度二甲基亚砜的研究》







 我要推广仪器
我要推广仪器
 下载APP
下载APP