给位大侠,我在根据国标法测奶粉中的牛磺酸时,配置乙酸锌溶液用来去蛋白,可是乙酸锌不溶于水。通过加热溶解时发现40℃时几乎没溶,后来水浴温度调到80℃时不停搅拌后还有一些未溶,但是温度再高的话里面的水又会蒸发了,于是又加了点凉水进去,随着温度的降低乙酸锌又被析出来了,放置一夜后烧杯上全是乙酸锌,而且烧杯里面乙酸锌又与原先加水时的量一样多,这是怎么回事?
做油过氧化值时,配置的乙酸异辛烷发生了分层现象。乙酸异辛烷的比例是按照国标GB/T 5538 中规定的3:2配置的。不知道大家有遇到过这种情况吗?是什么原因导致分层呢?
各位大神,哪有有二正辛基-双(巯乙酸2-乙基己酯)(DOTE)和三(2-乙基己基巯基乙酸)辛锡(MOTE)反应物料,或者有卖三(2-乙基己基巯基乙酸)辛锡(MOTE)的供应商啊。网上百度,相关信息为零。这是SVHC中的一项物质。http://simg.instrument.com.cn/bbs/images/default/em09512.gif
配置流动相,没有氯化锌,可以用乙酸锌代替吗

http://ng1.17img.cn/bbsfiles/images/2015/06/201506170935_550426_2770921_3.jpg乙酸锌-乙酸钠配置出来是浑浊的,想问问大家配出来是怎么样的,求图
大家好,我急求用岛津气相色谱分离,乙酸和乙酸乙酯的分离条件,柱子,进样口,检测器温度,以及乙醇和乙酸乙酯的分离条件,柱子采用RTX-17ms,急求,谢谢大家!!!
5750.10-2023新国标二氯乙酸和三氯乙酸的结果计算公式,不打算用方法给的公式,还有其他推出来的公式吗,求指教[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307042211154108_8493_6020871_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307042211155038_7049_6020871_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307042211155782_3893_6020871_3.png[/img]
如何定性及定量过氧乙酸和过氧辛酸,有过氧乙酸对照品,无过氧辛酸对照品.两种物质都比较容易分解.有经验或有方法的发出来共享一下.谢谢大侠!
提前感谢各位赐教。看氰化物的标准时发现,5750中氰化物(异烟酸-吡唑酮法)蒸馏时使用的是乙酸锌溶液+固体酒石酸,而484氰化物(异烟酸-吡唑啉酮)中用到的则是硝酸锌溶液和酒石酸溶液。想问一下这两者的的区别在哪里呢?日常操作中是否可以用乙酸锌溶液代替硝酸锌溶液呢?
想请教一下各位老师,为什么我用C18柱检测一针乙酸乙酯的时候,为什么乙酸乙酯在2min就出峰了,相似相容的话,乙酸乙酯应该最迟出峰啊?
要分析乙酸中的甲酸和乙酸乙酯,TCD检测器,填充柱。 用GDX加酸不太好分离,请教诸位同仁有无经验?
各位老师,乙酰乙酸乙酯说有烯醇类和酮类平衡的混合物。那是不是就是说,我进一针乙酰乙酸乙酯的分析纯,应该出两个峰,一个乙酰乙酸乙酯(烯醇类),一个是乙酯乙酯乙酯(酮类)。
谁告诉我下 巯基乙酸异辛酯 二甲基二氯锡 用GC什么检测器?什么柱子分析啊?
[color=#444444]GC-2014C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做标准曲线,TCD(热导)检测器。标准溶液是水、乙醇、乙酸和乙酸乙酯的混合物,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]出的图中,水合乙醇的峰很好,但乙酸和乙酸乙酯的峰有问题,乙酸的峰拖尾严重,乙酸乙酯的峰很小(质量分数和水差不多,但峰面积比水差很多,有事几乎没有)。进样器140℃,柱温120℃,检测器130℃(以前三个温度分别是175,140,175但是这种条件下乙酸乙酯量很少时15%以下,不出峰)。请问有没有办法是避免乙酸峰拖尾太严重,另外增加乙酸乙酯的峰面积?[/color]
新国标5750.10中的二氯乙酸回收率为什么很高,超出回收率范围,求大佬指教
我在GDX-102气象色谱柱(热导池法)上测混合物(含乙酸。乙醇。正丙醇。异丁醇。异戊醇。乙酸乙酯。乙酸正丙酯。乙酸异丁酯。乙酸异戊酯),请问专家:怎样控制气象色谱的操作条件能把这些混合物各自的含量测出来,而且峰形较好
各位老师:HPLC色谱条件下进样乙酸乙酯,分别在磷酸二氢钾(ph3.0)和甲酸流动相体系下乙酸乙酯的响应值相差较大,是否有同样的问题出现?
乙酸锌和亚铁氰化钾反应方程式怎么书写,?[color=#333333]亚铁氰化钾可配合乙酸锌作为澄清剂:它是利用乙酸锌与亚铁氰化钾反应生成的氰亚铁酸锌沉淀来挟走或吸附干扰物质。这种澄清剂除蛋白质能力强,但脱色能力差,适用于色泽较浅,蛋白质含量较高的样液的澄清,如乳制品、豆制品等,可以用于可溶性糖类的提取和澄清。[/color][color=#333333][/color][color=#333333]百度上有人这么回答,我想知道具体的反应方程式,或者[color=#333333]氰亚铁酸锌的分子式怎么书写?[/color][/color]
配制流动相时有的加一定比例的乙酸,通常的比例是按冰乙酸还是乙酸(36%)加入?两者的浓度相差很大,以至最终流动相的pH不一样,大家是按哪个浓度啊?
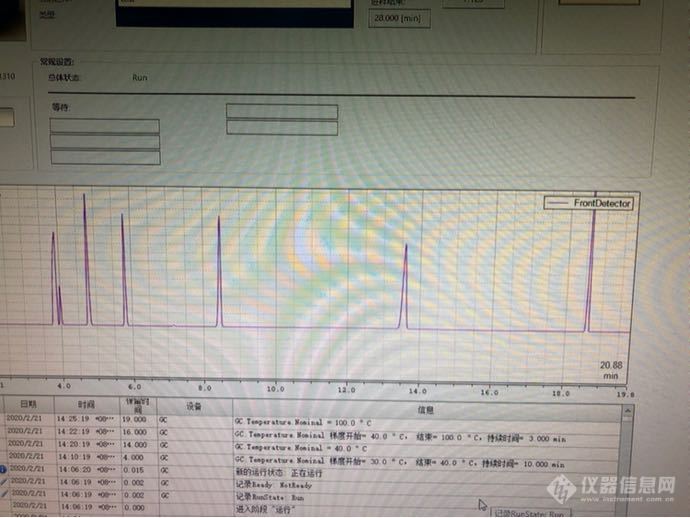
我买的坛墨的7种饱和脂肪酸酯类化合物混标!甲酸甲酯 乙酯 乙酸甲乙丙丁戊酯。现在问题来了。我甲酸乙酯和乙酸甲酯一直分不开 。下面是我条件:30 度保持4min 以1 度/min 升到40度 30度/min到100。后面都没问题 甲酸甲酯我温度低一点也能和cs2分开。可是甲酸乙酯和乙酸甲酯...分不开。那我怎么做呢?是不是要单独做一个乙酸甲酯曲线方法验证。柱子是 ffap 30 0.32 0.25[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/02/202002211425479329_4319_2990176_3.png[/img]
用茚三酮法测定氨基酸总量的时候先要配制乙酸-乙酸钠缓冲液,实验指导派介绍的是先称45.5克乙酸纳溶于100ml水中,加热溶解蒸发至60ml,冷却后加30ml乙酸,在用水定容于100ml容量瓶中.但我们在具体操作中却怎么也做不到,先是加热够冷却,但发现烧杯壁上附着厚厚一层药品,并且容量瓶里的溶液很快变成了糨糊,30ml乙酸和10ml的水根本冲不干净,误差太大.第二次干脆在还温着的时候就倒进容量瓶,但效果还是和第一次差不多.第三次是加热后趁热加乙酸进去,但还是那样.不知道别人做的时候有没有这种情况,是怎么解决的?我急啊~~~~~~~~~~~~~~~~~~```[em16] [em49]
哪位大侠有这些标准:SH/T 1628.1—1996 工业用乙酸乙烯酯SH/T 1628.2—1996 工业用乙酸乙烯酯纯度及有机杂质的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法SH/T 1628.3—1996 工业用乙酸乙烯酯活性度的测定 发泡法SH/T 1628.4—1996 工业用乙酯乙烯酯酸度的测定 滴定法SH/T 1628.5—1996 工业用乙酯乙烯酯中醛含量的测定 容量法
[color=#444444]有没有人知道怎么将乙醇,乙酸,乙酸乙酯,乙醛,水分开的方法,高水相,试着用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分,可是检测到的信号太弱,不识别,液相色谱分,流动相又是稀硫酸,有没有人用过其他办法,谢谢[/color]
求助:乙酸-乙酸镁的配制方法
昨天做实验发现 往浓硫酸加入乙酸乙酯时 没有分层而是乙酸乙酯全部溶到浓硫酸里面。这时什么原因呀?浓硫酸能氧化乙酸乙酯吗?请高手指教?谢谢!
SH/T 1628.1—1996 工业用乙酸乙烯酯SH/T 1628.2—1996 工业用乙酸乙烯酯纯度及有机杂质的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法SH/T 1628.3—1996 工业用乙酸乙烯酯活性度的测定 发泡法SH/T 1628.4—1996 工业用乙酯乙烯酯酸度的测定 滴定法SH/T 1628.5—1996 工业用乙酯乙烯酯中醛含量的测定 容量法
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url],氟苯尼考项目,用5%氨化乙酸乙酯作为提取液,涡旋,振荡,离心后取上清液氮吹,有时候会出现白色晶体,有时候又没有,请问下:1、氨水和乙酸乙酯会反应吗?2、5%氨化的乙酸乙酯会出现分层,刚配制好也会分层,那加上清液的时候,需要混匀后再加吗?还是只加上层3、5%氨化的乙酸乙酯需要现用现配吗?
目前我有一个体系中含有甲醇,乙酸,乙酸甲酯,以及少量的硫酸,我想问怎么才能够准确的知道甲醇,乙酸,乙酸甲酯的量,另外气相在进样过程中如何避免甲醇和乙酸之间的反应
气相色谱分离工业乙酸乙酯的条件??
大家好: 请问三氟乙酸乙酯中三氟乙酸含量该如何检测?1、如果用化学滴定,应怎么操作2、如果选用GC,该选什么色谱柱,需用外标法吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP