第一次做水质苯胺类化合物11889-1989里面标曲绘制要求每个比色管加50mg硫酸氢钾我想咨询线这硫酸氢钾起的作用是调节ph在1。5-2之间嘛?有没有替代方法?感觉每次称量很麻烦
如题,请问校准试验邻苯二甲酸氢钾标准溶液里要加硫酸汞吗?
大家好。我想问一下。 我的做1,4-对氨基苯与FITC(异硫氰酸荧光素)反应的时候。其条件完全是按照蛋白质的氨基与FITC 的条件。即氨基与异硫氰根的反应条件进行的。但是却标记不上。不知道是不是苯环上的氨基具有惰性?还是反应条件不适合?有哪位高手指教一下!谢谢啊 同时我还想问一下氨基和异硫氰基的反应原理及条件?
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
请问有谁做过二氯苯.二甲苯.苯的残留溶剂.请指教 采用什么柱子,和什么方法
[em06] 四氢呋喃、二氧六环、吡啶、甲苯 照残留溶剂测定法(附录Ⅷ P第三法)试验。精密称取苯适量,加甲醇制成每1 ml中约含60μg的溶液,作为内标溶液。精密称取四氢呋喃、二氧六环、吡啶、甲苯适量,加甲醇制成每1ml中各含720μg、380μg、200μg和890μg的溶液,作为对照贮备溶液;精密量取对照贮备溶液1ml与内标溶液1ml,置10ml量瓶中,加水稀释至刻度,摇匀,作为对照溶液。精密称取本品1.0g,置10ml量瓶中,加内标溶液1ml,加水溶解并稀释至刻度,摇匀,作为供试品溶液。用二乙烯基-乙基乙烯苯型高分子小球作为固定相,柱温190℃,依法测定。残留溶剂含量应符合规定。我让色谱公司按这个要求做了不锈钢柱子(柱填料:401有机载体(二乙烯基苯/乙基乙烯苯共聚体)60-80目),可是不出峰,后来把柱寄回去了,现在又寄给我的柱子(柱填料:10%PEG-20M CHROMOSORB PAW-DMCS 80-100目),峰是有了,可是分不开,我做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的氮气4圈,空气4.2圈,氢气4.5圈,后我又把氮气开到3圈,还是这个样子.是怎么回事呢,请高手赐教.谢谢!!!
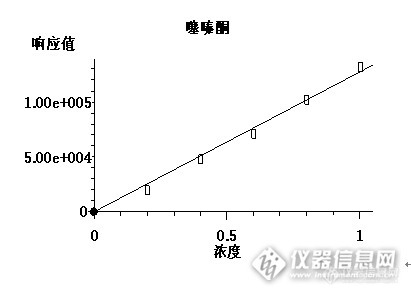
茶叶中硫丹、甲氰菊酯、噻嗪酮类农药残留的检测1、仪器: 安捷伦5975C/7890GC-MS;电子分析天平,箱式电阻炉,涡旋混合器;固相萃取装置;氮吹仪;安谱 GraphiCarb 固相萃取小柱(57084;250mg/3mL)。2试剂:硫丹、甲氰菊酯、噻嗪酮混合标准溶液;无水硫酸钠、色谱纯乙腈、甲苯;茶叶样品硫丹、甲氰菊酯、噻嗪酮混合标准使用液的配制:(α-硫丹、β-硫丹,硫丹硫酸盐、甲氰菊酯、噻嗪酮)0、0.2、0.4、0.6、0.8、1.0ug/mL。http://ng1.17img.cn/bbsfiles/images/2014/12/201412270910_529372_2266096_3.png3、样品准备将1g粉碎茶叶加到50mL离心试管中,加入3mL水润湿,浸泡10min,加15mL色谱纯乙腈,涡旋2min,静置,过滤,滤液待用,再用15mL色谱纯乙腈重复提取一次,过滤,合并滤液,滤液用50度的水浴中氮吹浓缩至2mL左右待净化。净化 柱子上端装入约1cm高的于650度灼烧过的无水硫酸钠层,以吸附除去多余的水分。a 活化: 4mL 乙腈:甲苯=3:1活化,流出液弃去;b 上样:将待净化的样液加入小柱(安谱 GraphiCarb 固相萃取小柱(57084;250mg/3mL)),收集流出液;用6mL 乙腈:甲苯=3:1分两次洗涤,合并流出液。c 重新溶解:于50度的水浴中氮吹浓缩至近干,加2mL农残级的正已烷溶解定容,GC-MS 检测。4、仪器条件色谱柱:HP-5MS; 30m*0.25mm*0.25um ;柱温:初始温度80 ºC,维持1min,以30 ºC/min 升温至200 ºC,并保持1min,再以5 ºC/min 升温至280 ºC,保持3 min;流速:1.0mL/min ;载气:高纯氦气;进样量:1μL;进样方式:不分流;进样口温度:230 ºC;接口温度:[
请问:我想分析 对三氯甲硫基苯甲酰氯的含量,用什么分析方法合适,可否用液相分析,采用什么柱子比较好?
求助无水硫酸镁,氢化钠,氯甲酸乙酯,四氢呋喃,乙腈(hplc),N-甲基吗啉,二异丙基乙胺,对甲苯磺酸的质量标准。大家有哪个给我哪个就可以。这些资料库里都没有。在外面也实在找不到了。大家帮忙。
我配制了一桶0.3mol/L氢氧化钠标准溶液,用基准物邻苯二甲酸氢钾标定出来的数据波动很大,根本不平行。可是用硫酸标准溶液标定出来却很平行(己排除了滴定管、基准物、烘箱的准确度因素),而且有个奇怪的现象,就是邻苯二甲酸氢钾标称样量一样数据就能平行,如果称样量不一样数据就不能平行,称样量越大标定出来的数据越高,称样量越小标定出来的数据就越低(比如称1.9g邻苯二甲酸氢钾标定得0.3050mol/L,称1.5g邻苯二甲酸氢钾标定得0.3000mol/L,如果8组都称1.9g邻苯二甲酸氢钾8组都标定得0.3050mol/L,如果8组都称1.5g邻苯二甲酸氢钾8组都标定得0.3000mol/L)这是为什么啊?
我配制了一桶20L的0.25mol/L氢氧化钠标注溶液,用邻苯二甲酸氢钾标定数据一点都不稳定,最高的数据0.2495,最低的0.2444,可是用硫酸标准溶液标定却平行得很好,为什么呀?不明白呀?知道的快快指导下吧,我快郁闷死了!
要标定氢氧化钠,在GB601标准中,是用邻苯二甲酸氢钾,但试剂库里找到苯二甲酸氢钾,请问大家,可以用吗?
最近在看GB5009.19-2008的方法,其中有一个农残 五氯苯基硫醚 从网上找不到CAS号,只能找到甲基五氯苯基硫醚,这两种物质是一样的吗?哪位大侠知道?
2012年3月15日,欧盟发布COMMISSION IMPLEMENTING REGULATION (EU) No 221/2012,修订对抗寄生虫剂/抗体内寄生物药剂氯氰碘柳胺Closantel的残留限量要求,新增对该兽药在乳【牛,绵羊】Milk中的临时残留限量要求45μg/kg,该临时残留限量将于2014年1月1日到期。该法规自公布3天后生效。 http://www.instrument.com.cn/news/20120316/075585.shtml 同一天欧盟发布COMMISSION IMPLEMENTING REGULATION (EU) No 222/2012,修订对抗寄生虫剂/抗体内寄生物药剂三氯苯哒唑Triclabendazole的残留限量要求,新增对该兽药在乳【所有反刍动物】Milk中的临时残留限量要求10μg/kg,该临时残留限量将于2014年1月1日到期。该法规自公布3天后生效。 http://www.instrument.com.cn/news/20120316/075585.shtml 欧盟在一次针对兽药上调检出标准。熟悉兽药检测的大侠们,检出量要求越来越精准,对检测仪器的有没有什么要求呢?这两种又是采用何种检测仪器和手段能够检测出。大家都有哪些话要说呢?
硫化氢的对氨基二甲基苯胺物质冻结成块了,这该怎么配?
刚接手做硫化物检测,方法是:n,n-二乙基对苯二胺分光光度法,方法说是稳定蓝色,可是加入显色剂后开始是蓝色然后马上变成乳白色浑浊了,做了多次都是这样,一直找不到原因,硫化物标准用的是国家标准溶液稀释的,请哪位兄弟帮忙解答下,多谢了,还有就是水样采来后加了乙酸锌,下面有很多白色浑浊沉淀,请问取水样该怎么取,有可能的话能否把硫化物显色照片发来看看
请问各位专家,水质苯系物的盲样是水中的苯系物还是二硫化碳中的苯系物,或者是甲醇中的苯系物?这三种到底是哪一种呢?如果是后两种,那么盲样还用萃取吗?依据国标是《水质苯系物的测定[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法》 采用的是二硫化碳萃取法。请各位专家指教!
哪位同道有 对氨基二甲基苯胺比色法 GB 18056 –2000 附录A 用来检测甲硫醇的,急需!!!
在测定不锈钢中铬时,加入指示剂N-苯基邻氨基苯甲酸,进行指示剂校正时,指示剂是消耗六价铬吗?那为什么GB223.4-2008滴定法测锰中,计算硫酸亚铁铵浓度时和最后计算锰浓度时都是减去校正值,我觉得应该加上才对啊,谁能解释解释?
我想请问大家哪些溶剂因为生产工艺中用到了苯,所以需要检测苯的残留的?甲醇乙醇肯定需要,还有吗?谢谢大家
跪求硫酸烟酰苯胺和吡啶-3-甲酰替苯胺的生产厂家!!!!!
测纯化水的硝酸盐时用0 .1%二苯胺硫酸液试剂,发现一周前配制的0 .1%二苯胺硫酸液试剂呈浅蓝色的已经变质,后来均是如此,现在只能现用现配,大家有没有更好的方法啊!
四氢呋喃、二氧六环、吡啶、甲苯 照残留溶剂测定法(附录Ⅷ P第三法)试验。精密称取苯适量,加甲醇制成每1 ml中约含60μg的溶液,作为内标溶液。精密称取四氢呋喃、二氧六环、吡啶、甲苯适量,加甲醇制成每1ml中各含720μg、380μg、200μg和890μg的溶液,作为对照贮备溶液;精密量取对照贮备溶液1ml与内标溶液1ml,置10ml量瓶中,加水稀释至刻度,摇匀,作为对照溶液。精密称取本品1.0g,置10ml量瓶中,加内标溶液1ml,加水溶解并稀释至刻度,摇匀,作为供试品溶液。用二乙烯基-乙基乙烯苯型高分子小球作为固定相,柱温190℃,依法测定。残留溶剂含量应符合规定。问题一:柱温190℃根本无法分离,甲醇峰还没走完 其他峰都出来了。是不是柱子太短了?用柱温190℃做出了的柱子长度是多少?问题二:3年前做过用程序升温,分离还可以,现在用同样的柱子同样条件做分离度不行了,柱子用了很久分离效果为什么会变差?那怎么办?问题三:查资料有报道说苯的浓度要大十倍。大十倍会好些吗?
本人用气相色谱做二硫化碳中的苯系物,买了很多厂家的二硫化碳(色谱纯)都不能用,杂峰太多,特别是在苯的出峰时间附近。提纯二硫化碳又太麻烦,而且毒性又大。各位同仁,知不知道哪个厂家的二硫化碳能用?请不吝赐教!
我分析苯系物时用到二硫化碳试剂萃取或解析样品,采用国标方法提纯国产二硫化碳的过程对人体毒害较大,原默克有无苯级二硫化碳试剂购买,较为方便。现默克不再卖此产品,请教哪位前辈知道哪有无苯级二硫化碳试剂购买,如没有,含苯量很低的二硫化碳试剂也可考虑!最好有具体的联系电话!谢谢!
用安捷伦7890――5977,做水中有机氯,百菌清,六氯苯,前处理为固相萃取,方法为生活饮用水卫生标准,现在的结果是666回收很稳定,ddt和六氯苯很差,哪位大师做过,请赐教。和
小弟做了好几次2-溴,3,3‘-二甲氧基联苯的氢谱,都发现甲基有三重峰,做何解?谢了!
按照空气和废气第四版亚甲蓝分光光度法测定硫化氢样品/空白加入混合显色剂后先变红色后慢慢颜色褪去,这个显色过程是不是有问题?我以前做样品显色的时候是浑浊变澄清后慢慢显红色或者蓝色,求教,可能是什么的影响此次过程中混合使用液重新配置:吸取2.5ml储备液,用(1+1)硫酸稀释至100ml 按照1.00ml对氨基二甲基苯胺溶液和1滴(0.04ml)三氯化铁溶液的比例混合
请教,分光光度法用试剂--对苯二胺(简称DPP)和硫酸高铁胺测定H2S时,此反应是否为硫化氢的特征反应,其他形式的硫化物有无响应?







 我要推广仪器
我要推广仪器
 下载APP
下载APP