有没有做过N-亚硝基胺的?我用N-二甲基亚硝基胺标准物质进样,但是根本观察不到信号,TIC和SIM都没有信号,这是什么原因呢?
大家谁有2-硝基-3甲基苯甲酸分析条件?谢谢
三甲基氢醌与三甲基苯醌之间的关系,怎么计算两者之间的含量
大家好,谁知道2-甲基-3-硝基苯甲酸的分析方法
亚硝基二甲胺和二甲基亚硝胺是不是一种东西?
2-甲基苯并咪唑和邻硝基苯胺的的紫外吸光度事多少?
我用滴定对苯醌的方法(溶液加碘化钾,盐酸,暗处静置后用硫代硫酸钠滴定)测三甲基苯醌含量,但是终点总是反色,找不到终点,请问高人们有解决的办法吗?谢谢!
[font=&][size=18px]顺序为:苯,萘,蒽,硝[/size][/font][font=&][size=18px]苯[/size][/font][font=&][size=18px]【用途】是染料、塑料、合成橡胶、合成树脂、合成纤维、合成药物和农药等的重要原料,也是涂料、橡胶、胶水等的溶剂,也可以作为燃料.[/size][/font][font=&][size=18px] 【制备或来源】工业上由焦煤气(煤气)和煤焦油的轻油部分提取和分馏而得.也可由环己烷脱氢或甲苯歧化或与二甲苯加氢脱甲基和蒸气脱甲基制取.[/size][/font][font=&][size=18px] 【其他】闪点10~12℃.蒸气与空气形成爆炸混合物,爆炸极限1.5%~8.0%(体积)[/size][/font][font=&][size=18px]萘[/size][/font][font=&][size=18px]工业上最重要的稠环芳烃.纯品为具有香樟木气味的白色晶体,熔点80.3℃.主要用于生产邻苯二甲酸酐、染料中间体、橡胶助剂和杀虫剂等.1958年以来,代替滴滴涕等氯化产品的甲萘威投产后,用作杀虫剂原料的比例有所增加.萘的用途分配,各国有所不同,大致用于生产邻苯二甲酸酐约占70%,染料中间体(如β-萘酚)和橡胶加工助剂约占15%,杀虫剂约占6%,鞣革剂约占4%,染料生产较少的国家,如美国则用于生产杀虫剂的比例较大.[/size][/font][font=&][size=18px]蒽[/size][/font][font=&][size=18px]用途 用作发光材料(如在闪烁计数器中),特别是用于涂层(如用于吸收紫外光).用于制造蒽醌和染料等.也用作杀虫剂、杀菌剂、汽油阻凝剂等.[/size][/font][font=&][size=18px] 制备或来源 在蒸馏煤焦油最后阶段得到,可由煤焦油的蒽油部分分出.[/size][/font]

气相色谱法对维生素E的原料三甲基氢醌的检测摘要 三甲基氢醌即2,3,5-三甲基氢醌,又名2,3,5-三甲基对苯二酚,是生产维生素E的中间体,其主要用途是用作生产维生素E的主要原料。目前,维生素E已成为国际市场上用途广泛、产销量极大的主要维生素品种,国内外市场前景广阔。目前全国生产能力不能满足国内市场供应不足,部分依赖进口。因此对三甲基氢醌的需求日益增加。而对于三甲基氢醌检测目前国家和行业都没有一个统一的检测标准。为此南京科捷分析仪器应用研究所根据客户的要求应用GC5890C气相色谱仪对2,3,5-三甲基氢醌进行方法研究。实验结果表明:本方法简便,分析速度快。能满足生产质量控制的要求,从而降价低生产成本。关键词 2.3.5- 三甲基氢醌 2,3,5-三甲基对苯二酚 维生素E中间体 气相色谱法一.2.3.5三甲基氢醌气相色谱图 http://ng1.17img.cn/bbsfiles/images/2011/06/201106171053_300271_2242538_3.jpg三、仪器配置 检测项目2,3,5-三甲基氢醌及其杂质色谱仪器型号GC5890C型色谱仪 配有FID检测器毛细管色谱柱0.32*30*0.25专用柱色谱工作站N2000(电脑1台自备)氮氢空发生器 HGT300E 1台或高纯氮、氢气、空气钢瓶各一瓶
求《HG/T 4415-2012 2,3,5-三甲基氢醌》标准,谢谢!
[b][size=4] 一、性状[/size][/b][size=4] 醌类化合物随着助色团酚羟基的引入而表现出一定的颜色。引入的助色团越多,颜色则越深。[/size][size=4] [b] 二、升华性[/b][/size][size=4] 游离的醌类多具升华性,小分子的苯醌类及萘醌类具有挥发性。[/size][b][size=4] 三、溶解性[/size][/b][size=4] 游离醌类多溶于有机溶剂,微溶或不溶于水。而醌类成苷后,极性增大。[/size][b][size=4] 四、酸碱性[/size][/b][size=4] 蒽醌类衍生物酸性强弱的排列顺序为:含COOH>含二个以上β-OH>含一个β-OH>含二个以上α-OH>含一个α-OH.在分离工作中,常采取碱梯度萃取法来分离蒽醌类化合物。用碱性不同的水溶液(5%碳酸氢钠溶液、5%碳酸钠溶液、1%氢氧化钠溶液、5%氢氧化钠溶液)依次提取,其结果为酸性较强的化合物(含COOH或二个β-OH)被碳酸氢钠提出;酸性较弱的化合物(含一个β-OH)被碳酸钠提出;酸性更弱的化合物(含二个或多个α-OH)只能被1%氢氧化钠提出;酸性最弱的化合物(含一个α-OH)则只能溶于5%氢氧化钠。[/size][b][size=4] 五、显色反应[/size][/b][size=4] (1)Feigl反应 醌类衍生物在碱性条件下加热与醛类、邻二硝基苯反应,生成紫色化合物。医学教育网搜集整理[/size][size=4] (2)无色亚甲蓝显色试验 无色亚甲蓝乙醇溶液(1mg/ml)专用于检识苯醌及萘醌。样品在白色背景下呈现出蓝色斑点,可与蒽醌类区别。[/size][size=4] (3)Borntrager's反应 在碱性溶液中,羟基醌类颜色改变并加深,多呈橙、红、紫红及蓝色,如羟基蒽醌类化合物遇碱显红至紫红色,称之为Borntrager's反应。蒽酚、蒽酮、二蒽酮类化合物需氧化形成羟基蒽醌后才能呈色,其机理是形成了共轭体系。[/size][size=4] (4)Kesting-Craven反应 当苯醌及萘醌类化合物的醌环上有未被取代的位置时,在碱性条件下与含活性次甲基试剂,如乙酰乙酸酯、丙二酸酯反应,呈蓝绿色或蓝紫色。蒽醌类化合物因不含有未取代的醌环,故不发生该反应,可用于与苯醌及萘醌类化合物区别。[/size][size=4] (5)与金属离子的反应 蒽醌类化合物如具有α-酚羟基或邻二酚羟基,则可与Pb[sup]2+[/sup]、Mg[sup]2+[/sup]等金属离子形成络合物。[/size][size=4] 与Pb[sup]2+[/sup]形成的络合物在一定pH条件下能沉淀析出,与Mg[sup]2+[/sup]形成的络合物具有一定的颜色,可用于鉴别。如果母核上只有1个α-OH或1个β-OH,或2个-0H不在同环上,则显橙黄至橙色;如已有1个α-OH,并另有1个-0H在邻位则显蓝至蓝紫色,若在间位则显橙红至红色,在对位则显紫红至紫色。[/size]
做食品中的总蒽醌,标准品用混合碱溶解后是紫红色,样品用酸水解后用三氯甲烷提取,最后用混合碱提取,得到的溶液是土黄色,色系不同怎么比色呢?有没有做过总蒽醌的老师告知一下如何检测总蒽醌
急求二甲基砜的检测方法,目前正在试[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]方法,想问下用SPB-5弱极性柱是否能检测二甲基砜,文献上用的中性柱,但实验室没有,选用丙酮做溶剂,硝基苯做内标,进样口柱温检测器温度都是190度,或者有除了[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]别的检测方法也可以
茶叶中蒽醌的测定解决方案蒽醌,是一种醌类化合物,欧盟认为其具有致癌性,将茶叶中蒽醌的限量标准定为0.02 mg/kg。我国是茶叶出口大国,输欧茶叶经历了年初唑虫酰胺农残项目屡遭欧盟通报退货的绿色壁垒后,近来欧盟又加大了对我国输欧茶叶中蒽醌残留项目的检测力度。截至2014年11月,某省已有8批茶叶遭欧盟通报退货,其中6批是唑虫酰胺超标,2批是蒽醌超标。茶叶中蒽醌问题已引起欧盟官方及我国茶叶行业的广泛关注。方法优势:目前有关蒽醌检测的文献及标准较少,迪马科技开发的《茶叶中蒽醌的测定》具有:采用固相萃取-GCMS法,用乙酸乙酯、正己烷提取,通过ProElut TPC净化, GCMS分析;能够达到准确定性定量,检出限为6 μg/kg,定量限为20 μg/kg,与欧盟给出的限量标准一致;前处理步骤简单、回收率高、方法稳定性好、净化效果优异等特点;特别适用于输欧茶叶中的蒽醌检测。以下为详细解决方案,敬请参考!茶叶中蒽醌的测定1、适用范围适用于茶叶中蒽醌的检测,方法检出限6 μg/kg,定量限20 μg/kg。2、样品准备称取5 g样品于离心管中,向离心管中加入20 mL乙酸乙酯,振荡2 min,6000 rpm下离心2 min,收集上层清液;向下层残渣中加入20 mL乙酸乙酯:正己烷=1:1按照步骤(1)提取一次,合并两次上清液;将上清液在35 ℃下减压蒸干,5 mL乙腈-甲苯*超声溶解,待净化。3、SPE柱净化——ProElut TPC(Cat.# 65354)(1)活 化:向柱中加入2 g无水硫酸钠,10 mL乙腈-甲苯*活化;(2)上 样:将待净化液加入小柱,弃去流出液;(3)淋 洗:向柱中加入10 mL乙腈-甲苯*,弃去流出液;(4)洗 脱:向柱中加入15 mL乙腈-甲苯*,收集流出液;(5)重新溶解:将洗脱液在40 ℃下减压蒸干,冷却,用正己烷定容至1 mL,供GCMS分析。*乙腈-甲苯溶液:乙腈:甲苯=3:1(体积比)4、色谱条件色谱柱:DM-5MS 30 m × 0.25 mm × 0.25 μm(Cat.# 8221)进样口温度:300 ℃升温程序:初始温度100 ℃,保持1 min,以10 ℃/min升温至280 ℃,保持5 min载气:氦气,流速:1.37 mL/min进样方式:不分流进样进样量:1 μL离子源温度:260 ℃接口温度:300 ℃溶剂延迟:2.9 min电子轰击电离源(EI):选择离子监测模式(SIM),分组监测见表1表1 选择离子监测组表通道起始时间结束时间选择离子(m/z)12.924152,180,2085、添加回收结果茶叶中蒽醌添加回收结果化合物名称添加水平(μg/kg)回收率(%)蒽醌20104.2http://www.dikma.com.cn/u/image/2016/02/01/1454313054900999.jpg 蒽醌标准(0.1 μg/mL)的(m/Z-152)GCMS图茶叶中蒽醌的测定相关产品信息:货号名称规格样品前处理65354茶叶J检测专用柱 ProElut TPC12 mL 20/pkg24435812管防交叉污染真空SPE萃取装置12位48031,3,6mL柱管通用连接器15/pk4806考克(控制流量)15/pk99011真空/正压两用泵,无油1/pk99013抽滤瓶套装(包括硅橡胶管2米,2L抽滤瓶及橡胶塞)1/pk30039FitMax针头式过滤器 Nylon13 mm,0.22 μm 100/pk30040FitMax针头式过滤器 Nylon13 mm,0.45 μm 100/pk标准品46581蒽醌100 mg色谱柱及保护柱8221DM-5MS30 m × 0.25 mm × 0.25 μmHPLC溶剂Ÿ缓冲盐Ÿ离子对试剂50104乙酸乙酯 HPLC级4 L50101乙腈 HPLC级4 L50115正己烷 HPLC级4 L通用色谱产品52401B瓶架/蓝色(现货)[td=1,1,12
采用保健食品标准与技术规范的方法进行总蒽醌的检测,先用酸水解2h,然后用三氯甲烷回流萃取,耗时且浪费CHCl3溶剂,有时候采用该方法检测原料时还不能分层,问题多多,为什么保健食品中总蒽醌的检测不能采用药典的方法呢?有过问一个专家,说是专家认可保健食品标准与技术规范中的方法。大家讨论一下你们实验室做总蒽醌时是怎么做的?
茶叶中蒽醌的测定解决方案蒽醌,是一种醌类化合物,欧盟认为其具有致癌性,将茶叶中蒽醌的限量标准定为0.02 mg/kg。我国是茶叶出口大国,输欧茶叶经历了年初唑虫酰胺农残项目屡遭欧盟通报退货的绿色壁垒后,近来欧盟又加大了对我国输欧茶叶中蒽醌残留项目的检测力度。截至2014年11月,某省已有8批茶叶遭欧盟通报退货,其中6批是唑虫酰胺超标,2批是蒽醌超标。茶叶中蒽醌问题已引起欧盟官方及我国茶叶行业的广泛关注。方法优势:目前有关蒽醌检测的文献及标准较少,迪马科技开发的《茶叶中蒽醌的测定》具有:采用固相萃取-GCMS法,用乙酸乙酯、正己烷提取,通过ProElut TPC净化, GCMS分析;能够达到准确定性定量,检出限为6 μg/kg,定量限为20 μg/kg,与欧盟给出的限量标准一致;前处理步骤简单、回收率高、方法稳定性好、净化效果优异等特点;特别适用于输欧茶叶中的蒽醌检测。以下为详细解决方案,敬请参考!茶叶中蒽醌的测定1、适用范围适用于茶叶中蒽醌的检测,方法检出限6 μg/kg,定量限20 μg/kg。2、样品准备称取5 g样品于离心管中,向离心管中加入20 mL乙酸乙酯,振荡2 min,6000 rpm下离心2 min,收集上层清液;向下层残渣中加入20 mL乙酸乙酯:正己烷=1:1按照步骤(1)提取一次,合并两次上清液;将上清液在35 ℃下减压蒸干,5 mL乙腈-甲苯*超声溶解,待净化。3、SPE柱净化——ProElut TPC(Cat.# 65354)(1)活 化:向柱中加入2 g无水硫酸钠,10 mL乙腈-甲苯*活化;(2)上 样:将待净化液加入小柱,弃去流出液;(3)淋 洗:向柱中加入10 mL乙腈-甲苯*,弃去流出液;(4)洗 脱:向柱中加入15 mL乙腈-甲苯*,收集流出液;(5)重新溶解:将洗脱液在40 ℃下减压蒸干,冷却,用正己烷定容至1 mL,供GCMS分析。*乙腈-甲苯溶液:乙腈:甲苯=3:1(体积比)4、色谱条件色谱柱:DM-5MS 30 m × 0.25 mm × 0.25 μm(Cat.# 8221)进样口温度:300 ℃升温程序:初始温度100 ℃,保持1 min,以10 ℃/min升温至280 ℃,保持5 min载气:氦气,流速:1.37 mL/min进样方式:不分流进样进样量:1 μL离子源温度:260 ℃接口温度:300 ℃溶剂延迟:2.9 min电子轰击电离源(EI):选择离子监测模式(SIM),分组监测见表1表1 选择离子监测组表通道起始时间结束时间选择离子(m/z)12.924152,180,2085、添加回收结果茶叶中蒽醌添加回收结果化合物名称添加水平(μg/kg)回收率(%)蒽醌20104.2http://www.dikma.com.cn/u/image/2016/02/01/1454313054900999.jpg 蒽醌标准(0.1 μg/mL)的(m/Z-152)GCMS图茶叶中蒽醌的测定相关产品信息:货号名称规格样品前处理65354茶叶J检测专用柱 ProElut TPC12 mL 20/pkg24435812管防交叉污染真空SPE萃取装置12位48031,3,6mL柱管通用连接器15/pk4806考克(控制流量)15/pk99011真空/正压两用泵,无油1/pk99013抽滤瓶套装(包括硅橡胶管2米,2L抽滤瓶及橡胶塞)1/pk30039FitMax针头式过滤器 Nylon13 mm,0.22 μm 100/pk30040FitMax针头式过滤器 Nylon13 mm,0.45 μm 100/pk标准品46581蒽醌100 mg色谱柱及保护柱8221DM-5MS30 m × 0.25 mm × 0.25 μmHPLC溶剂Ÿ缓冲盐Ÿ离子对试剂50104乙酸乙酯 HPLC级4 L50101乙腈 HPLC级4 L50115正己烷 HPLC级4 L通用色谱产品52401B瓶架/蓝色(现货)50孔5240
最近,茶叶等作物中发现一种新的污染物残留--蒽醌。这蒽醌是什么家伙,有什么危害和特点,怎么检测?欢迎知道的网友来讨论。
液质联用---蒽醌类,有两种是蒽醌,另一种是二苯并吡喃环,同时测定三种物质,使用的负离子模式,两种蒽醌类相应比较高,他们的结构也相似,另一种的相应很低,用甲酸水时会使蒽醌类拖尾减轻,但是另一种会有抑制的作用,响应变低,如果用乙酸铵,蒽醌会有拖尾现象严重,另一种会响应好一点,这三种响应都不是很高,响应低的在1000ng时才几十的峰面积,另两个蒽醌是在500ng时有 1000多的峰面积,我想请问,什么因素影响响应高低?流动相会很大影响物质的响应?是不是我单标条件摸错了,子离子母离子fragment等等,,,像我这种一种响应很低的该怎么办?最大的可能是什么呢?是该怀疑我之前的摸条件错了吗?还是把研究的重点放到流动相的问题上?
想做一个熔点标准物,现在手里有蒽醌粗品,如何把它纯化?请指教!!!
最近扩项皮革N亚硝基胺 GB/t24153-2009 5ppm 12种混标打下去, N-亚硝基吡咯烷,N-亚硝基N-甲基苯胺,N-亚硝基乙基苯胺 ,N-亚硝基二苯基胺都没有,查找离子发现有N-甲基苯胺,N-乙基苯胺,二苯胺,推测热分解。那么N-亚硝基吡咯烷应当分解为吡咯烷才对,然而附近并没有找到对应峰......手边没有N-亚硝基吡咯烷单标,不好验证。有没有经验丰富的老师讲解一下,这种情况是不是热分解导致的,N-亚硝基吡咯烷是不是也分解了呢?如果分解了,产物是什么,特征离子多少。还是其他原因造成的?进样口温度260 质谱280 DB-35柱子 分段升温38-300
[color=#444444]现在确定里面有2-乙基蒽醌236和四氢-2乙基蒽醌240,可是没找到,找到了238和242的,是什么原因[/color][color=#444444][img]http://muchongimg.xmcimg.com/data/bcs/2016/0301/w123h4065258_1456817910_472.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/data/bcs/2016/0301/w201h4065258_1456817917_827.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/data/bcs/2016/0301/w127h4065258_1456817926_200.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/data/bcs/2016/0301/w197h4065258_1456817932_818.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/data/bcs/2016/0301/w126h4065258_1456817940_678.png[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/data/bcs/2016/0301/w200h4065258_1456817945_175.png[/img][/color][color=#444444][/color][color=#444444][/color]
[color=#231815]大黄提取物中5种蒽醌化合物的分离纯化[/color][color=#231815][color=#333333]为研究大孔树脂对大黄5种蒽醌的分离效果,本文采用静态吸附实验,比较6种大孔树脂(HPD-100、XDA-6、AB-8、LX-38、ADS-7和ADS-17)对5种游离蒽醌(芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚)的吸附及解吸附性能,筛选出对大黄5种蒽醌吸附率和吸附率最高的大孔树脂。然后以筛选的大孔树脂作为载体,对其动态吸附特性进行了初步研究。结果显示,HPD-100大孔树脂对大黄5种蒽醌吸附率和吸附率最高 在层析柱径高比1∶8,上样溶液5种蒽醌总浓度为3.64 mg/mL,上样体积2.0 BV,流速1.0 BV/h,85%的乙醇洗脱,洗脱体积为3.0 BV的优化条件下,HPD-100对5种蒽醌的动态吸附率为86.3%,洗脱率为85.9%,5种蒽醌总含量增加了2.88倍,由原来的7.13%增加到20.5%,总回收率98.7%,提取物中残留的离子液体Br也同时被除去,表明本实验选择的优化条件具有可行性。[/color][/color]
[b]问题:[b][/b]13种-N-亚硝胺混标,适用于《GB/T 28482-2012婴幼儿安抚奶嘴安全要求》、《EN-71-12-71122013玩具安全第12部分亚硝胺和亚硝基类物质共12种》化合物有哪些?答案:1.N-亚硝基吡咯烷2.N-亚硝基二甲胺3.N-亚硝基吗啉4.N-亚硝基二乙胺5.N-亚硝基哌啶6.N-亚硝基-N-甲基苯胺7.N-亚硝基二异丙胺8.N-亚硝基二丙胺9.N-亚硝基-N-乙基苯胺10.N-亚硝基二异丁胺11.N-亚硝基二丁胺12.N-亚硝基二苄胺13.N-亚硝基-二乙醇胺[/b][align=center]=======================================================================[/align]【[b]活动内容[/b]】1、每个工作日上午10:00左右发布一个色谱问答题,版友根据题目给出自己理解的答案。2、每个工作日下午15:10公布参考答案。【[b]活动奖励[/b]】[b]幸运奖:[/b]抽奖软件,当天随机抽取3个或5个回答正确的版友ID号(最后一个ID号,截止至下午15:00),每人奖励[b][color=#ff0000]2钻石币[/color][/b](抽奖人数≤10,抽取3个版友;抽奖人数>10,抽取5个版友);[b]幸运奖5名(2钻石币)[/b]风云变幻(注册ID:v3165605)zengzhengce163(注册ID:zengzhengce163)ZHAOGUANGXI(注册ID:ZHAOGUANGXI)mengzhaocheng(注册ID:mengzhaocheng)PAEs(注册ID:v2911392)[img=,690,502]http://ng1.17img.cn/bbsfiles/images/2018/02/201802091509265888_7664_708_3.jpg!w690x502.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2018/02/201802091509403422_4656_708_3.jpg!w690x388.jpg[/img][align=left][color=#ff0000][b]PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。[/b][/color][/align][align=left][color=#ff0000][b] 下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。[/b][/color][/align]
本人需要用HPLC 分析蒽醌的含量,有知道的老师给介绍一下,谢谢啊![em03]
样品制备 制备方法:标准品衍生方法:取硝基呋喃代谢物混标200 μL,加入2 mL 0.2 mol/L 盐酸溶液和20μL 衍生剂,混合后置于37℃条件下保持4 h。注:0.2 mol/L 盐酸溶液:量取17 mL浓盐酸,用水定容至1000 mL。衍生剂:含2-硝基苯甲醛0.05 mol/L。称取0.075 g 2-硝基苯甲醛溶于10 mL二甲基亚砜,现用现配。分析条件 色谱柱:EndeavorsilTM C18 ,100×2.1 mm,1.8 μm(Cat#:87003) 流动相:A: 0.4% 甲酸水B:乙腈流速:0.2 mL/min柱温:30 ℃检测器:UV 254 nm进样量:2.0 μL梯度洗脱表时间(min)A:0.4%甲酸水B:乙腈07030370303.0120808.0020808.01703015.007030http://www.dikma.com.cn/Public/Uploads/images/2(112).PNGhttp://www.dikma.com.cn/Public/Uploads/images/3(89).PNG
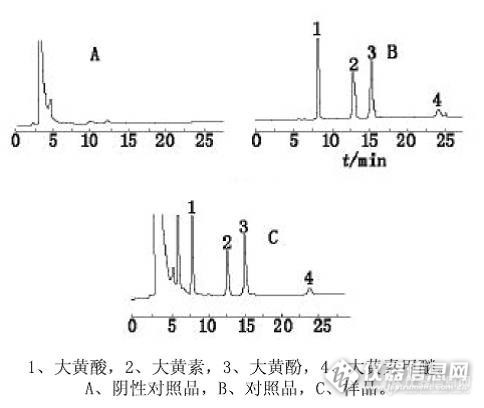
大黄总蒽醌来源于掌叶大黄的根茎,其颜色形状为棕黑色浸膏或棕色粉状结晶,是游离蒽醌、结合蒽醌、蒽酮、糖、鞣质等混合物,生物活性与大黄原药相同。对大黄进行研究之后,从大黄之中分离得到了此类化合物,药理研究表明,此类化合物具有泻下的作用。现在的中药提取物,都是采用一定的方法对药材进行提取,大黄中富含此类成分,提取得到的就为此类化合物的混合物,也就称为总蒽醌。以下为使用资生堂色谱柱对大黄药材检测得到的谱图,请参考。http://ng1.17img.cn/bbsfiles/images/2016/11/201611240934_01_2222981_3.jpg【色谱条件】色谱柱:CAPCELL PAK C18 S5; 4.6 mm i.d.×250 mm流动相:甲醇/0.1%磷酸溶液=70/30(原条件:甲醇/0.1%磷酸溶液=85/15)流 速:1.0mL/min温 度:30°C检 测:UV254nm进样量:20μL*摘自:解放军药学学报,2009年2月,第25卷,第1期,52-55
请问各位高人,有没有从事关于1.4-二羟基蒽醌检测的,分析它常用的化学分析方法是什么?用液相分析的条件是什么样的?谢谢
请问专家:我急需要做黑索今(三次甲基三硝胺,一种六元环形化合物的高能炸药,环上带3个硝基)的粒度分布,用的分散剂是二辛基琥珀酸磺酸钠,分散介质用的是水,平行结果不好,请问有没有什么合适的分散剂和分散介质?-我用的是美国库尔特公司的LS-230激光粒度仪。——盼回复,不胜感激!
蒽醌(相对分子质量208)初始浓度10ppm(原来实验初始浓度0.5ppm),降解24h,流动相是甲酸水溶液,乙腈,乙腈从1%洗到90%,正离子和负离子各出一个比较明显的峰(已和空白流动相和0min样品做对比),现不清楚该如何做质谱谱图解析,求教大神指点!图1是负离子模式下,明显的峰有258 226 220 97图2是正离子模式下,明显的峰有171 101目前分析:97是苯甲基的峰,226是邻甲苯某酯,正离子模式分析不出来,但是226也不确定是邻甲苯某酯,求教大神该如何分析[img]https://ng1.17img.cn/bbsfiles/images/2018/10/201810191059427733_6411_3487194_3.png[/img][img=,690,1225]https://ng1.17img.cn/bbsfiles/images/2018/10/201810191100462555_5228_3487194_3.png[/img][img=,690,1225]https://ng1.17img.cn/bbsfiles/images/2018/10/201810191101001853_7802_3487194_3.png[/img]
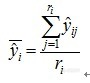
[align=center][b]基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的2,3,5-三甲基苯醌粗品萃取过程定量模型优化研究[/b][/align][b]中文摘要:目的[/b]实际工业生产工艺中,萃取是一项耗时耗力的过程,萃取终点的确定通常采用离线的HPLC, [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]或由熟练工人根据经验判断,这些方法操作较复杂或是不够准确,在实际生产中缺乏一种快速有效的检测手段以判断萃取终点,节省操作时间,避免过分萃取浪费溶剂。利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术可以明显改善萃取工艺。[b]方法[/b]本实验针对2,3,5-三甲基苯醌(TMBQ)粗品萃取环节,采用偏最小二乘法(PLS)建立模型,考察了不同预处理方法与变量选择方法对模型的影响以优化模型,采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]结合PLS算法建立TMBQ萃取过程含量快速检测模型,并使用不同预处理方法与波段选择方法对模型进行优化,最终确定使用一阶导数+SG15点平滑预处理结合iPLS选择波段建立PLS模型。[b]结果[/b]建立模型的各项参数为:波普区间4385.33cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup], 5928.11cm[sup]-1[/sup]-6309.94cm[sup]-1[/sup],模型决定系数R[sup]2[/sup]=0.996, RMSEP=0.1350。[b]结论[/b]建立的模型精密度与准确度良好,可以满足含量分析的需要,是TMBQ萃取过程含量快速检测的有效方法,可以快速准确的对三甲基苯醌粗品萃取过程进行在线监测,提供了一种用于该工艺环节的快速检测手段,如果应用于生产,可以节省操作时间,避免溶剂浪费。[b]关键词:[/b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析;2,3,5-三甲基苯醌;萃取 2,3,5-三甲基苯醌是维生素E的主要中间体。2,3,5-三甲基苯醌在国外已有生产, 但国内尚未见文献报道。国内用2,3,5-三甲基苯醌主要依赖进口。因此,开展2,3,5-三甲基苯醌的合成研究对发展国内维生素 E 的生产具有重要意义。TMHQ的合成工艺国内外己有多种报道,较为先进的是TMP法与异佛尔酮法,TN[b]B[/b]Q粗品萃取过程是合成TMBQ的关键环节。在制药领域,NIRS作为一种重要的PAT工具,已成功用于药物的原辅料评价、关键过程的监测和控制、成品的快速放行和质量监测等各个环节,为保证产品质量、降低生产成本、革新生产过程发挥了重要的作用。[b]1实验材料与仪器1.1仪器[/b] Antaris Ⅱ傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国Thermo Fisher公司),7890A[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]-氢焰离子化检测器(美国Agilent公司),HP-1毛细管色谱柱(美国Agilent公司)BT224S电子分析天平(德国Sartorius公司),容量瓶,100ml圆底烧瓶,分液漏斗,[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url][/color][/url](美国ThermoFisher公司)。RESULT[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]采集软件,TQAnalyst[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析软件,Matlab数据处理软件。[b]1.2试剂[/b] 2,3,6-三甲基苯醌(合成步骤见第二章),石油醚(天津富宇精细化工有限公司,沸程60℃-90℃)。[b]2方法2.1样品制备和处理[/b] 按照第二章步骤合成得TMBQ得其石油醚溶液,萃取水相合并有机相,旋蒸浓缩除去石油醚至橙黄色油状液体,称重,再用石油醚作为溶剂配置1ug/ml~50mg/ml一系列溶液。[b]2.2光谱采集[/b] 波长范围4000 cm[sup]-1[/sup]-10000cm[sup]-1[/sup];扫描次数32;分辨率8 cm[sup]-1[/sup],使用4mm光程的玻璃样品管乘装液体样品,采集样品前采集背景以消除背景干扰,每个样品重复采集三次光谱。光谱采集在恒定室温(24℃)与恒定湿度的条件下进行。[b]2.3样品集划分[/b] 使用K-S分类法将所有66个样品换分为48个校正集与18个验证集。[b]2.4模型建立与优化[/b] 采用导数、平滑等方法对原始光谱进行预处理,应用偏最小二乘法(PLS)建立模型,结合RMSEP等评价参数,通过变量选择方法选择特征波段优化模型。[b]2.5 重复性考察[/b] 选择3个验证集样品,每个样品连续采集10次光谱,使用建立好的模型预测每张光谱,并计算出每个样品十次预测值的均值和标准偏差。是第i个样品的第j张光谱,第i个样品共测定ri个光谱,第i个样品的预测平均值为:[align=center][img=,90,83]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311044_01_1626619_3.png[/img][/align] 复测定的标准偏差为:[align=center][img=,164,102]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311044_02_1626619_3.png[/img][/align] 用c[sup]2[/sup]检验来考察这些重复性标准偏差是否属于同一总体:[align=center][img=,271,245]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311045_01_1626619_3.png[/img][/align] z为需要重复测定的样品数,将所得χ[sup]2[/sup]与自由度(z-1)临界值比较,若χ[sup]2[/sup]在临界值以下,则重复测定的所有方差属于同一总体,标准偏差均值σ可以作为近红外测定的标准偏差,近红外分析方法的重复性为z××σ[sub]max[/sub]。如果χ[sup]2[/sup]大于临界值,近红外分析方法的重复性随样品组分浓度不同而不同,这时,近红外分析方法的重复性不大于z××σ[sub]max[/sub](σ[sub]max[/sub]为σi中的最大值)。[b]2.6[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测[/b] 初始温度180℃恒温5min,以10℃/min的速率升温至240℃。进样口温度:300,检测器温度:300,载气:氮气,载气流速:3ml/min,进样量:0.5ul。[b]3结果3.1校正集与验证计划分[/b] 使用K-S分类法将所有66个样品换分为48个校正集与18个验证集。校正集与验证集的第一第二主成分分布图如图1,其中黑色符号代表校正集样品,红色符号代表验证集样品,验证集均匀分布于校正集中,可见使用该方法分类合理。[align=center][img=,553,217]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311047_01_1626619_3.png[/img][/align][align=center]图1 所有样品主成分分布图[/align][b]3.2预处理方法的选择[/b] 考察无预处理、一阶导数+SG5点平滑、一阶导数加SG9点平滑、一阶导数+SG15点平滑、二阶导数加15点平滑这几种方式的建模结果,以RMSEC、RMSECV、RMSEP以及R[sup]2[/sup]作为评价指标,结果见表1。[align=center]表1 预处理方法评价参数[/align][align=center][img=,566,164]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311104_01_1626619_3.png[/img][/align] 无预处理的模型结果最差,说明噪声对模型结果有较大影响,原始光谱如图2。SG15点平滑+一阶导数的预处理结果RMSEC、RMSECV以及RMSEP最小,R[sup]2[/sup]最高。因此选择SG15点平滑+一阶导数作为模型的预处理方法,预处理后光谱如图3。[align=center][img=,524,224]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311048_01_1626619_3.png[/img][/align][align=center]图2 原始光谱图[/align][align=center][img=,532,210]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311049_01_1626619_3.png[/img][/align][align=center]图3 一阶导数+SG15点平滑预处理光谱图[/align][b]3.2异常样本的剔除[/b] 图4为校正集样品在学生残差-杠杆值图中的分布。图中5号(红色方框标记)样品学生残差值与杠杆值都非常高,判定为异常样品,猜测为溶液配制错误或者在光谱采集过程中出现错误,因此在后期模型优化中剔除这一异常值。[align=center][img=,563,217]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311050_01_1626619_3.png[/img][/align][align=center]图4 学生残差-杠杆值关系图[/align][b]3.3波段选择结果[/b] 以一阶导数+SG15点平滑为最优预处理方法进行波段选择,主要考察ForwardiPLS、SPA、相关系数法三种方法。[b]3.3.1iPLS波段选择结果[/b] 设定20为最大主成分数,分别考察以50、100、200个变量为波段基础的建模效果。红色虚线是全波段建模的RMSECV,红色与绿色条带的高度代表以此条带的变量建模所得RMSECV,从图5中可见,绿色条带的RMSECV值最小,因此绿色条带是被选择用于建模的波段,红色条带则表示不被选择的区域。表2为各变量基础的模型参数。[align=center][img=,558,268]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311051_01_1626619_3.png[/img][/align][align=center]图5 以50个变量为基础的iPLS法波段选择效果图[/align][align=center][img=,572,266]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311052_01_1626619_3.png[/img][/align][align=center]图6 以100个变量为基础的iPLS法波段选择效果图[/align][align=center][img=,618,262]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311052_02_1626619_3.png[/img][/align][align=center]图7 以200个变量为基础的iPLS法波段选择效果图[/align][align=center]表2 不同变量基础的建模结果[/align][align=center][img=,646,111]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311053_01_1626619_3.png[/img][/align][b]3.3.2 SPA法波段选择结果[/b] SPA算法首先通过完成n个波长分组各M个波长选择,然后通过多元定量校正模型完成m(1£m£M)个最优波长的选定。图8为SPA法选择变量的效果图。 运行SPA算法共选择3个变量,对应波数为4188.65cm[sup]-1[/sup],4885.50cm[sup]-1[/sup],7503.50cm[sup]-1[/sup],为图中红色方框标注,以此3个变量建立PLS模型,结果如表 所示,RMSECV与RMSEP均有所增加,R[sup]2[/sup]降低,表明模型预测能力与线性都有所降低。分析原因可能是此方法在选择波段过程中由1557个变量减少到3个,光谱变量删除过多,去除大量无关变量的同时导致许多有价值信息的丢失。[align=center][img=,501,246]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311053_02_1626619_3.png[/img][/align][align=center]图8 SPA算法变量选择结果图[/align][b]3.3.3相关系数法波段选择结果[/b] 将相关系数阈值设定为0.6、0.7、0.8,使用相关系数法计算出TMBQ含量值与波数的相关系数图,如图9,图中虚线为设定的相关系数阈值,虚线以上及以及的部分代表相关系数大于阈值的波段,阈值越高,被选择的波段越少,当阈值设为0.8时,大于阈值的波段已经较少。以超过阈值的波段建立PLS模型。模型结果如表3,可见将阈值设为0.6时模型结果最好。[align=center] a[img=,402,175]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311055_01_1626619_3.png[/img][/align][align=center] b[img=,409,187]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311056_01_1626619_3.png[/img][/align][align=center] c[img=,409,176]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311056_02_1626619_3.png[/img][/align][align=center]图9 不同阈值的波数相关图(a阈值设为0.6,b阈值设为0.7,c阈值设为0.8)[/align][align=center]表3 相关系数法建模参数[/align][align=center][img=,496,105]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311058_01_1626619_3.png[/img][/align][b]3.4 小结[/b] 综合比较全波段建模与三种波段选择方法建模结果,参数如表。其中使用iPLS法选取600个变量,波段区间为4385.33cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup],5928.11cm[sup]-1[/sup]-6309.94 cm[sup]-1[/sup],分别对应双键上C-H第一组合频与一级倍频吸收,建模后具有最高的决定系数和最低的各项方差值,这些参数表明使用该方法建立的模型预测能力最好,与真实值最接近。因此本实验主要选择iPLS方法选择变量,结合一阶导数+SG15点平滑建立模型,应用于TMBQ萃取过程含量的快速检测。[align=center]表4 各变量选择方法比较[/align][align=center] [img=,374,136]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311059_01_1626619_3.png[/img][/align][align=center][img=,524,214]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311059_02_1626619_3.png[/img][/align][align=center]图10 优化后模型预测线性图[/align][b]3.5重复性试验考察[/b] 采集验证集8号、25号、36号样品,对TMBQ含量模型进行重复性测试,每样品采集10次光谱。预测结果见表5。[align=center]表5 重复性考察结果[/align][align=center][img=,578,337]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311100_01_1626619_3.png[/img][/align] 自由度为2时,χ[sup]2[/sup]临界值为5.99。实际χ[sup]2[/sup]小于临界值,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析方法重复性为0.154,可以满足分析应用。[b]3.6NIR预测考察[/b] 第一次使用20ml石油醚萃取,之后每次使用等体积10ml石油醚萃取,共萃取8次,使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]测定TMBQ峰面积,并使用NIR采集8次萃取液[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url],使用优化好的定量模型对其含量进行预测。[align=center][img=,490,255]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311102_01_1626619_3.png[/img][/align][align=center]图11 NIR预测值[/align] 图11为NIR对萃取过程的预测结果,第一次萃取即将大部分产品萃取出,随后的每次萃取量呈逐渐下降的趋势,在第五次萃取后,萃取液中产品含量几乎为0,并且随后没有变化,表明已达到萃取终点。使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测第4~8次萃取液,记录TMBQ峰面积,结果如表6。[align=center]表6 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测结果表[/align][align=center][img=,529,66]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311103_01_1626619_3.png[/img][/align] 第五次萃取后,TMBQ峰面积已经很小,并且基本没有变化,因此在4次萃取完全可以将水相中的TMBQ萃取完全,继续萃取已经没有意义,[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测与NIR预测结果相符,表明此模型预测能力良好,对萃取工艺具有一定指导意义。[b]4讨论[/b] 本实验采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]结合PLS算法建立TMBQ萃取过程含量快速检测模型,并使用不同预处理方法与波段选择方法对模型进行优化,最终确定使用一阶导数+SG15点平滑预处理结合iPLS选择波段建立PLS模型,建模所用波段区间为4385.33 cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup],5928.11 cm[sup]-1[/sup]-6309.94cm[sup]-1[/sup],模型决定系数R[sup]2[/sup]=0.996,RMSEP=0.1350。使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]验证了NIR模型对萃取过程与终点的预测能力。以上结果表明模型精密度与准确度良好,可以满足含量分析的需要,是TMBQ萃取过程含量快速检测的有效方法。[b]5参考文献[/b]孙月婷. 维生素E 的合成与分析研究现状. 广州化工, 2011, 39(6): 34-35.O.A.Kholdeava Synthesis of Vitamia E J.Mol.Cotal,1992,88(5):235~ 244孔黎明, 周涛, 菅盘铭. 2, 3, 5- 三甲基苯醌和2, 3, 5- 三甲基氢醌的一种合成方法: 中国, 102219665. 2011-10-19.A BShishmakov, Yu V Mikushina, O V Koryakova. Oxidation of 2,3,6-Trimethylphenolon Titanium Dioxide Xerogel by Hydrogen Peroxide in the Absence of an OrganicSolvent. RUSSIAN JOURNAL OF APPLIED CHEMISTRY, 2011, 84(9):1555-1559. O V Zalomaeva, N N Trukhan,I D Ivanchikova, et al. EPR study on the mechanism of H[b][sub]2[/sub][/b]O[b][sub]2[/sub][/b]-basedoxidation of alkylphenols over titanium single-site catalysts. J. Mol.Catal. A: Chem., 2007, 277(1-2), 185~192.褚小立. 化学计量学方法与分子光谱分析技术.北京 化学工业出版社. 2011.董学锋,戴连奎,黄承伟等.结合PLS-DA与SVM的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]软测量方法







 我要推广仪器
我要推广仪器
 下载APP
下载APP