求助:托吡酯和果糖二丙酮的质量标准(右完整分析方法)
求分离一氯丙酮、1,3-二氯丙酮、1,1-二氯丙酮、1,1,3-三氯丙酮、1,1,1-三氯丙酮、1,1,1,3-四氯丙酮的GC分析方法?
在做土VOC时,数据分析处理时发现丙酮,二氯甲烷和二硫化碳值都很大,主要是丙酮和二氯甲烷,有几百万那么大。做土svoc和voc时,是分房间处理的,就算交叉污染也不应该值这么大吧……纯水高我可以理解,没加内标和替代,但实验室空白和样品值那么大,我就无法理解。离子源液洗过了,再做一遍值有几万了,但还是太大。二氯甲烷可能跟我这儿的纯水机有关,我这儿用的不是屈臣氏,但丙酮那么大我就一点头绪都没有了……请问问有没有做土水气voc的大神解答一下,谢谢??![img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907220838277197_6211_3862253_3.png[/img]
做蔬菜样品时,先加的水和丙酮,加NaCl分层后,上层应该是丙酮吧?但为什么开始加的丙酮比水多很多,分层后丙酮却比水相少很多呢?而且色素全在丙酮里。 分出来的水相再加二氯甲烷提,分层后下层是二氯甲烷吗?怎么也有人说加了NaCl使二氯甲烷在水的上层呢?
本人是一分析方面的菜鸟,有一问题向各位请教。我之前用HPLC分析水溶液中的葡萄糖,果糖,HMF等物质,所使用的柱子是Shodex SH-1011柱(分析糖类的柱子),流动相用0.5mM的硫酸溶液,柱温60度。由于现在实验的需要,待分析溶液中加入了高含量的有机溶剂(丙酮,DMSO等,以下以丙酮为例,含量高达90%),我现在还想用原来的那套系统进行分析,将柱温和检测器温度都调到50度以下,此外还需要注意哪些问题呢,比如是否需要用水将样品稀释还是可以直接进样等等。在此先拜谢各位了。
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.

大家好: 按照15版药典,检测阿拉伯胶的“葡萄糖和果糖”项目,结果是硅胶板在喷了显色剂,然后放入烘箱加热后,整个硅胶板都变黑了。.且后面研究发现,直接将显色剂喷到板上,板在烘箱中加热,板就变黑了。求助有此经验的同学,问题会出在哪里?我们自己调查的结果可能是如下几个方面,:1- 硅胶板质量问题(用了2个品牌的板,国药集团和上海信宜的,都出现了这样的问题,可能性小);2- 显色剂有问题(用的都是新开瓶的试剂,可能性小);3- 药典的方法有问题,要求用的硅胶G板,是不是应当用不同的板?附:检验方法葡萄糖和果糖 取本品0.1g ,置离心管中,加1%三氟乙酸溶液2 m l ,强力振摇使溶解,密塞120°C加热1 小时,离心,小心转移上层液至50ml烧杯中,加水10ml减压蒸发至干. 残渣加水0 .1m l及甲醇0.9ml,离心分离沉淀。如有必要,用醇1ml稀释上层清液。另分别取阿拉伯糖、半乳糖、葡萄糖、鼠李糖及木糖对照品各lOmg于lm l水中,用甲醇稀释至10ml,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各10μl,分别点于同一硅胶G 薄层板上,以1 .6%磷酸二氢钠溶液-正丁醇-丙酮(10:4 0: 50)为展开剂,展开,取出,晾干,喷以对甲氧基苯甲醛溶液(取对甲氧基苯甲醛0.5ml,加冰醋酸10m丨,甲醇8 5 m l,琉酸5ml,摇匀,即得)至恰好湿润,立即在110C加热10分钟,放冷,立即检视,对照品溶液应显示的5个淸晰分离的斑点,从下到上的顺序依次为半乳糖(灰绿色或绿色)、葡萄糖(灰色)、阿拉伯糖(黄绿色)、木糖(绿灰色或黄灰色)、鼠李糖(黄绿色)。供试品色谱中,在与半乳糖和阿拉伯糖对照品色谱相应的位置之间,不得显灰色或灰绿色斑点。http://ng1.17img.cn/bbsfiles/images/2015/10/201510130951_569848_1835550_3.jpg
测果糖中的二氨基苯乙酮,标准曲线好做吗?为什么我的老是不出峰,且基线不是很稳.紫外220nm,1.0ml/min,柱子C1840度.进样量是300ul很大的,各位有谁做过,给支个招吧谢谢
环己烷、正己烷、二氯甲烷、丙酮对PAH的萃取能力排序是怎么样的呢?另外,怎么样直观的判断一种溶剂对PAH的萃取能力呢?向大家求教了[em09511]
各位老师好:请问,老师有检测过药物中异丙叉丙酮和二丙酮醇痕量残留的分析的经验么?请指教。谢谢
我做的一个课题样品有100g,是二氯甲烷层,极性较小样品二氯甲烷溶解,硅胶拌样,干法装了个大柱子,规格为9cmx50cm我用小硅胶板做小试,试了各种溶剂系统如石油醚-乙酸乙酯,环己烷-丙酮,二氯甲烷甲醇最后发现环己烷-丙酮系统效果最好此系统的各种比例下,样品点都很圆,一串薄层斑点就像冰糖葫芦,很漂亮所以我决定用环己烷-丙酮来进行梯度洗脱柱子已经装好了,我发现个问题。。。。。。环己烷的密度0.779,丙酮的密度0.7899本来想通过测密度的方式进行回收溶剂的比例再调配二者密度太接近了,怎么办?或者有没有别的方法?希望大家积极出谋划策能有效解决问题的给予1-5个积分奖励~
附件是原料巯基丙酮用酒精稀释后进的gcms,请问巯基丙酮二聚体的峰到底是14.866还是22.072,或者说两者都是?还有,根据香料通则,这个东西的含量要达到95%,根据图上看有个很大的巯基丙酮,含量应该不到95%,巯基丙酮是本来就有的呢还是二聚体分解出来的?大家做原料控制的时候怎么做的呢?
请问一下买的丙酮中的环氧氯丙烷标准品,可以用丙酮做溶剂稀释吗,要用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]操作,标准里是用二氯甲烷做溶剂
我做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测棉铃虫体内的高效氯氰菊酯量,经过丙酮--石油醚提取浓缩后,用丙酮溶解,然后就是进[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测了,但是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]坏了,修好还需要一段时间,我的样品怎样保存啊,能保存多少时间?????各位大哥大姐给个答案吧, 谢谢啦!!!
中药农残检测样品前处理中为何要除尽丙酮及二氯甲烷/有没有人知道/
各位,有没有人做过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定单丙酮葡萄糖的含量(峰面积归一法)
求助:在做土壤半挥发性有机物中二氯甲烷和丙酮1:1的萃取体系有没有替代溶剂?
二氯甲烷,丙酮,正己烷,四氢弗喃这些试剂都是实验室长期使用的,危害程度如何?操作需要注意哪些
三溴丙酮和二溴丙酮要用什么色谱柱分离啊
三氯丙酮的性质?
请教老师:我想将乙醇、丙酮、乙酸甲酯、二异丙基胺、吡啶、二氯甲烷分开,您上次告知了柱温及方法,可是进样口温度、检测器温度、流速及分流比该怎么设定呢?多谢!
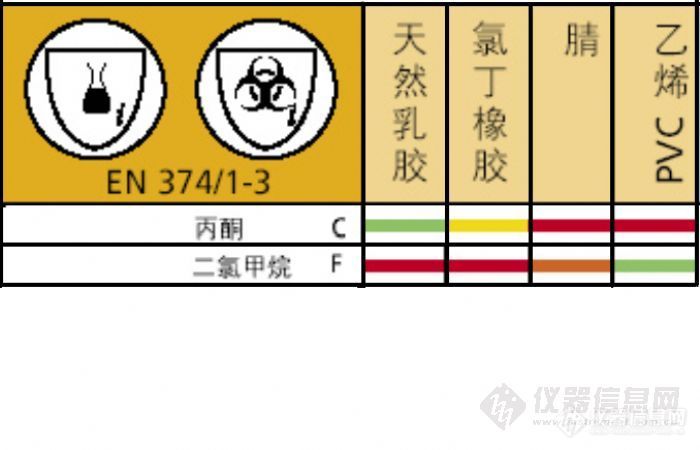
有的实验需要用到丙酮和二氯甲烷的混合溶剂,需要耐受这两种溶剂的手套,查找常用手套材料的溶剂耐受表,发现没有合适的,大家知道有哪种材料的手套符合要求?
本人现急需氯亚铂酸钾和二(乙酰丙酮)铂的分析方法或行业标准,那位大虾有相关资料,请上传或发到我的信箱[color=#DC143C]zhangfy03@126.com[/color],不胜感激!!!!
前段时间我们用NPD检测器,HP-5色谱柱,测有机磷样品,当时想用丙酮做溶剂,有的老师提出丙酮对色谱柱的影响太大,不让用丙酮,后来选择的二氯甲烷,虽然二氯甲烷对NPD也有影响,但我们还是这样做下来了。我只是想在此请教下专家,丙酮对柱子的影响大吗?以后都要尽量避免用丙酮吗?
我想将乙醇、丙酮、乙酸甲酯、二异丙基胺、吡啶、二氯甲烷分开,需要使用何种[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱,柱温、进样口温度、检测器温度、流速及分流比是怎样的?多谢!急急急!!!
我想净化我处理后的样品,按照丙酮正己烷活化,洗脱,但是购买丙酮还得备案,学校弄这个时间太长了,来不及。求助大家有什么可以替代丙酮的么?异丙醇或者是甲醇,二氯甲烷等。
做一氯丙酮的时候,使用方法90度恒温,一氯丙酮出峰时间在2.0左右,在2.9分有一个峰怀疑是一氯丙酮缩合物,网上查不出资料,有没有知道2.9分那个峰的沸点是多少,最好有具体详细的物性介绍
想请问一下各位高手,在丙酮和正己烷重蒸过程中,重蒸装置上放温度计的地方需要个塞子,我用的是橡胶塞,考虑到橡胶中肽酸脂会污染正己烷,从而对有机氯农药的测定带来影响,我想用铝箔纸把橡胶塞包起来,避免影响,但是不知道铝箔与正己烷和丙酮蒸汽接触时会不会带来其他的影响,想请问一下高手们这种处理方法行吗?或者有更好的处理方法?谢谢!
请问如何定量分析?二丙酮醇分解温度较低,请问要选用什么样的溶剂作内标物呢?另外我在单独进样二丙酮醇时,一直会出现两个峰,请问如何解决?我们公司二丙酮醇的供应商拿来的检测报告上面写着纯度为99.8%,有谁知道他们是如何测出来的,而且上面的谱图是一个峰.可是我怎么做都是两个峰,请问有更好的条件分离吗?谢谢...急!~~
有谁知道一氯丙酮的含量方法啊?查了写资料是用气相,具体怎么做呢?有谁知道标准呢,类似国标或者行标呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP