[b][/b][align=center][b]银黄颗粒质量标志物评价研究[/b][/align][b] 摘要[/b]目的:以黄芩药材、金银花药材、黄芩提取物、金银花提取物、银黄制剂为研究对象,考察并优化了样本在前处理环节的回流提取溶剂的体积、回流提取时间和提取溶剂的温度等。方法:采用高效液相色谱法,色谱柱为Venusil MP C[sub]18[/sub](4.6mm × 250 mm,5μm), Venusil MP C[sub]18[/sub](4.6mm × 250 mm,3μm)和 Agela MP S/N。以乙腈一0.3% 磷酸溶液为流动相进行梯度洗脱,流速为0.7 mLmin[sup]-1[/sup],检测波长为235 nm。结果和结论:通过各方面的考察,确定了银黄颗粒、黄芩药材和金银花药材在样品前处理环节的工艺优化参数,为银黄颗粒质量标志物研究提供借鉴指导。结论: 建立的提取方法稳定、可靠,有效成分达到最大提取效率,可用于银黄颗粒溯源检测的质量控制和综合评价。[b] 关键词:[/b]银黄颗粒;质量标志物;高效液相;黄芩;金银花[b] [/b][align=center][b][color=#333333]Evaluation of Quality Markers of Yinhuang Granules[/color][/b][/align]Objective: To investigate and optimize the volume ofreflux solvent, reflux extraction time and temperature of extraction solvent inthe pretreatment of samples, taking Scutellaria baicalensis, honeysuckle,Scutellaria baicalensis extract, honeysuckle extract and Yinhuang preparationas research objects. METHODS: High performance liquid chromatography was usedwith Venusil MP C18 (4.6 mm *250 mm, 5 micron), Venusil MP C18 (4.6 mm *250 mm,3 micron) and Agela MP S/N as chromatographic columns. The gradient elution was carried out with acetonitrile-0.3% phosphoric acid solution as mobile phase.The flow rate was 0.7 mL/min and the detection wavelength was 235nm. RESULTS AND CONCLUSION: The process optimization parameters of Yinhuanggranules, Radix Scutellariae baicalensis and Flos Lonicerae in samplepretreatment were determined through various aspects of investigation, whichcould provide reference and guidance for the study of quality markers ofYinhuang granules. CONCLUSION: The established extraction method is stable andreliable, and the effective ingredients can reach the maximum extractionefficiency. It can be used for quality control and comprehensive evaluation oftraceability detection of Yinhuang granules.Keywords: Yinhuang granules quality markers high performance liquidchromatography Scutellaria baicalensis honeysuckle[b]一、前言[/b] 银黄颗粒组方由金银花和黄芩构成,具有清热疏风、利咽解毒的功效,用于外感风热、肺胃热盛所致的咽干、咽痛、喉核肿大、口渴、发热急慢性扁桃体炎、急慢性咽炎、上呼吸道感染等症。该复方原料金银花为忍冬科植物忍冬的干燥花蕾或带初开的花,主产于山东、河南和河北等地。该复方原料黄芩为唇形科[url=https://baike.baidu.com/item/%E9%BB%84%E8%8A%A9%E5%B1%9E][color=windowtext]黄芩属[/color][/url]多年生草本植物,产于河北,河南,陕西,山西,山东等地。黄芩提取物的主要活性成分为黄芩苷、汉黄芩苷、黄芩素及汉黄芩素,金银花提取物是从金银花中提取的有机酸类活性成分。该制剂及其原料药成分复杂,生产厂家及产地众多,样品存在差异。中药质量标志物(Q-marker)已广泛应用于中成药的质量评价与控制。近年来越来越多的研究使用不同种类的分析仪器,密切联系中药有效性-物质基础- Q-marker研究,建立了丰富的中成药系统质量控制方法,为探讨建立中药全过程质量控制及质量溯源体系奠定了基础。[b]二、材料与方法1仪器与试剂、试药1.1仪器[/b] Waters e2695高效液相色谱仪(美国Waters公司),Waters 2998紫外检测器(美国Waters公司),Waters Empower色谱工作站(美国Waters公司);AGBP210S电子天平(Sartorius公司);MILLIPORE纯水机(MILLIPORE公司);高速万能粉碎机(北京市永光明医疗仪器有限公司,FW-80型);SB4200DTS超声波双频清洗机(宁波新芝生物科技股份有限公司);KDM-A控温电热套(金坛市医疗仪器厂);Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,5 μm)和Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,3 μm)。[b]1.2 试剂与试药[/b] 乙腈(上海星可高纯溶剂有限公司,色谱纯);甲醇(天津市科密欧化学试剂有限公司,色谱纯);其余试剂均为分析纯,水为超纯水。对照品来源:葛根素(批号:110752-200912)购自中国食品药品检定研究院。2样品的收集与前处理[b]2.1样品的收集[/b] 本研究从全国范围内收集黄芩、金银花药材各50批,分别制备相应的黄芩提取物和金银花提取物各50批,并制备银黄颗粒样品至少50批。(共计不少于250批样品)。[b]2.2黄芩、金银花药材的处理[/b] 对收集到的各批样品,均按照《中国药典》2015年版(第四部)药材取样法,四分法取样,1/4留样,剩余药材粉碎,使粉末分别过60目和20目筛,并按比例称重。所有黄芩、金银花药材样品均装袋密封,保存于冰柜(-20℃)中,备用。[b]2.3黄芩提取物的制备[/b] 取黄芩约100 g,置于1000 ml容量瓶中,加热回流两次,每次2 h,将滤液置于烧杯中浓缩至200 ml,用2 mol/L的盐酸调PH至1.0-2.0,80 ℃保温1 h,静置24 h.减压抽滤,沉淀加一倍量水混匀,用40 %氢氧化钠调节PH至7.0,加等量乙醇,搅拌溶解,滤过,滤液用2 mol/L的盐酸调PH 1.0-2.0, 60 ℃保温1 h,静置24 h,滤过,沉淀物加水洗至PH 5.0,95%乙醇洗至中性,挥尽乙醇,干燥,即得。[align=center]表1 黄芩提取物的提取[/align][align=center][img=,579,348]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091021374879_6392_3255306_3.png!w579x348.jpg[/img][/align][b]2.4金银花提取物的制备[/b] 称取金银花50.05 g置于圆底烧瓶中,加纯水回流提取三次,第一次8倍量水400 ml回流提取1 h,滤过,残渣加8 倍量水400 ml二次回流提取1 h,滤过,合并煎液,残渣加6倍量水300 ml,合并煎液,浓缩成浸膏,加浸膏量50%的淀粉混匀,置于烘箱中,60 ℃干燥,粉碎成粉,即得。[align=center]表2 金银花提取物的提取[/align][align=center] [img=,552,347]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091021550706_5584_3255306_3.png!w552x347.jpg[/img][/align][b]3供试品溶液方法考察的制备3.1供试品溶液制备方法考察3.1.1提取溶剂的选择[/b] 根据银黄颗粒的服用说明,该样品采用水为溶媒制备供试品溶液,由于临床应用中黄芩,金银花多采用水煎内服的用法,因此研究中以水作为提取溶媒,制备样品溶液。[b]3.1.2内标物溶液的制备[/b] 经查阅大量文献,本实验适用的内标物为葛根素。取葛根素对照品适量精密称定,以水超声溶解并定容制成浓度为30 μg*mL[sup]-1[/sup]的内标溶液[b]3.2银黄颗粒供试液制备方法考察3.2.1银黄颗粒不同料液比的考察[/b] 银黄颗粒研细后精密称取细粉1.0 g,称四份,置于100 ml或250 ml的圆底烧瓶中,分别精密加入煮沸的蒸馏水25 ml、50 ml、100 ml、150 ml于圆底烧瓶中,称重,加热回流30 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后0.5 ml等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表3、图1。[align=center]表3 银黄颗粒不同料液比单位质量色谱峰面积比较[/align][align=center] [img=,289,425]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091023281381_1955_3255306_3.png!w289x425.jpg[/img][/align][align=center]图1 银黄颗粒不同料液比单位质量色谱峰面积比较[/align][align=center][img=,289,123]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091023402582_6283_3255306_3.png!w289x123.jpg[/img][/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取体积为100 ml时值最大,最终选择回流提取体积为100 ml。[b]3.2.2银黄颗粒不同提取时间的考察[/b] 银黄颗粒研细后精密称取细粉1.0 g,称四份分别置于100 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水25 ml于圆底烧瓶中,称重,分别加热回流20 min、30 min、40 min、60 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后0.5 ml等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表4、图2。[align=center]表4 银黄颗粒不同提取时间单位质量色谱峰面积比较[/align][align=center] [img=,292,421]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091024007921_8570_3255306_3.png!w292x421.jpg[/img][/align][align=center][img=,577,251]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091024513811_6890_3255306_3.png!w577x251.jpg[/img][/align][align=center]图2 银黄颗粒不同提取时间单位质量色谱峰面积比较[/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取时间为30 min时值最大,最终选择回流提取时间为30 min。[b]3.2.3银黄颗粒冷热水的考察[/b] 银黄颗粒研细后精密称取细粉1.0 g,称两份分别置于250 ml的圆底烧瓶中,第一份精密加入煮沸的蒸馏水100 ml于圆底烧瓶中,第二份精密加入100 ml常温蒸馏水,称重,加热回流30 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表5、图3。[align=center]表5 银黄颗粒冷热水单位质量色谱峰面积比较[/align][align=center][img=,287,427]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091025490101_4911_3255306_3.png!w287x427.jpg[/img][/align][align=center][img=,574,270]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091026003331_9248_3255306_3.png!w574x270.jpg[/img][/align][align=center]图3 银黄颗粒冷热水单位质量色谱峰面积比较[/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取溶剂为热水时值最大,最终选择提取溶剂为热水。[b]3.3黄芩药材供试液制备方法考察 3.3.1黄芩药材不同料液比的考察[/b] 按比例称取2 0~60目和过60目筛的黄芩药材粉末,共0.57 g,称取三份,置于100 ml或250 ml的圆底烧瓶中,分别精密加入煮沸的蒸馏水25 ml、50 ml、100 ml于圆底烧瓶中,称重,加热回流30 min ,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表6、图4。[align=center]表6 黄芩药材不同料液比单位质量色谱峰面积比较[/align][align=center][img=,291,425]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091026251557_1424_3255306_3.png!w291x425.jpg[/img][/align][align=center][img=,567,260]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091026352391_8450_3255306_3.png!w567x260.jpg[/img][/align][align=center]图4 黄芩药材不同料液比单位质量色谱峰面积比较[/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取体积为50ml时值最大,最终选择50ml为最佳提取容积。[b]3.3.2黄芩药材不同提取时间的考察[/b] 按比例称取2 0~60目和过60目筛的黄芩药材粉末,共0.57 g,称取四份,分别置于100 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水25 ml于圆底烧瓶中,称重,分别加热回流20 min、30 min、40 min、60 min ,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表7、图5。[align=center]表7 黄芩药材不同提取时间单位质量色谱峰面积比较[/align][align=center][img=,289,424]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091027053867_9448_3255306_3.png!w289x424.jpg[/img][/align][align=center][img=,605,240]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091027125561_9333_3255306_3.png!w605x240.jpg[/img][/align][align=center]图5 黄芩药材不同提取时间单位质量色谱峰面积比较[/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取时间为40 min时值最大,最终选择40 min为最佳提取时间。[b]3.3.3黄芩药材冷热水提取的考察[/b] 按比例称取2 0~60目和过60目筛的黄芩药材粉末,共0.57 g,称取二份,置于100 ml的圆底烧瓶中,第一份精密加入煮沸的蒸馏水50 ml于圆底烧瓶中,第二份精密加入50 ml常温蒸馏水,分别称重,加热回流40 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表8、图6.[align=center]表8 黄芩药材冷热水单位质量色谱峰面积比较[/align][align=center][img=,287,424]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091027401011_8981_3255306_3.png!w287x424.jpg[/img][/align][align=center][img=,619,293]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091027478961_1794_3255306_3.png!w619x293.jpg[/img][/align][align=center][/align][align=center]图6 黄芩药材冷热水单位质量色谱峰面积比较[/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取溶剂为热水时值最大,最终选择热水提取是最佳的。[b]3.4金银花药材供试液制备方法考察3.4.1金银花药材不同料液比的考察[/b] 按比例称取2 0~60目和过60目筛的金银花药材粉末,共0.5 g,称取三份,置于100 ml或250 ml的圆底烧瓶中,分别精密加入煮沸的蒸馏水50 ml、100 ml、200 ml于圆底烧瓶中,称重,加热回流30 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表9、图7。[align=center]表9 金银花药材不同料液比单位质量色谱峰面积比较[/align][align=center][img=,358,511]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091031346401_7706_3255306_3.png!w358x511.jpg[/img][/align][align=center][img=,636,256]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091031349941_1562_3255306_3.png!w636x256.jpg[/img][/align][align=center]图7 金银花药材不同料液比单位质量色谱峰面积比较[/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取容积为100 ml,200 ml时值较大,100 ml与200 ml比较,两者的成分含量差别不大,所以选择100 ml提取较好。[b]3.4.2金银花药材不同提取时间的考察[/b] 按比例称取2 0~60目和过60目筛的黄芩药材粉末,共0.5 g,称取四份,分别置于250 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水100 ml于圆底烧瓶中,称重,分别加热回流20 min、30 min、40 min、60 min ,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算各共有峰的单位质量的峰面积值,比较其差异,结果见表10、图8。[align=center]表10 金银花药材不同提取时间单位质量色谱峰面积比较[/align][align=center][img=,290,424]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091030005792_6136_3255306_3.png!w290x424.jpg[/img][/align][align=center][img=,555,215]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091030053727_8053_3255306_3.png!w555x215.jpg[/img][/align][align=center][/align][align=center]图 9 金银花药材冷热水提取单位质量色谱峰面积比较[/align] 由上述表图分析:各主要共有峰的单位质量峰面积在提取溶剂为热水时值最大,最终选择热水提取最佳。[b]3.5 黄芩提取物,金银花提取物供试液制备方法考察[/b] 通过实验得知黄芩提取物,金银花提取物供试液制备方法考察同银黄颗粒供试液制备方法考察一致。[b]3.6 含葛根素内标的银黄颗粒、黄芩药材、金银花药材、黄芩提取物、金银花提取物供试液配制方法的确定3.6.1含葛根素内标银黄颗粒供试品溶液的配制[/b] 银黄颗粒研细后精密称取细粉1.0 g,置于250 ml圆底烧瓶内,精密加入煮沸的蒸馏水100 ml,称重,加热回流30 min(提前打开电热套预热),放冷,补重,过滤,取续滤液。另取葛根素对照品适量精密称定,以水超声溶解并定容制成浓度为30μg• mL-1的内标溶液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液。[b]3.6.2含葛根素内标黄芩药材供试品溶液的配制[/b] 从冰柜中取出黄芩药材粉末,放置室温。采用四分法取样,按比例称取2 0~60目和过60目筛的黄芩药材粉末,共0.57 g,置于100 ml圆底烧瓶中,精密加入煮沸的蒸馏水50 ml,称重,加热回流40 min(提前打开电热套预热),放冷,补重,过滤,取续滤液。另取葛根素对照品适量精密称定,以水超声溶解并定容制成浓度为30 μg• mL-1的内标溶液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液。[b]3.6.3含葛根素内标金银花药材供试品溶液的配制[/b] 从冰柜中取出金银花药材粉末,放置室温。采用四分法取样,按比例称取2 0~60目和过60目筛的黄芩药材粉末,共0.25 g,置于100 ml圆底烧瓶中,精密加入煮沸的蒸馏水50 ml,称重,加热回流30 min(提前打开电热套预热),放冷,补重,过滤,取续滤液。另取葛根素对照品适量精密称定,以水超声溶解并定容制成浓度为30 μg• mL-1的内标溶液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液。[b]3.6.4含葛根素内标金银花提取物供试品溶液的配制[/b] 从冰柜中取出金银花提取物粉末,按比例精密称取0.2 g,置于250 ml圆底烧瓶中,精密加入煮沸的蒸馏水100 ml,称重,加热回流30min(提前打开电热套预热),放冷,补重,过滤,取续滤液。另取葛根素对照品适量精密称定,以水超声溶解并定容制成浓度为30 μg• mL-1的内标溶液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液。[b]3.6.5含葛根素内标黄芩提取物供试品溶液的配制[/b] 从冰柜中取出黄芩提取物粉末,按精密称取0.04 g,置于250 ml圆底烧瓶中,精密加入煮沸的蒸馏水100 ml,称重,加热回流30 min(提前打开电热套预热),放冷,补重,过滤,取续滤液。另取葛根素对照品适量精密称定,以水超声溶解并定容制成浓度为30 μg• mL-1的内标溶液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液。[b]三、结论[/b] 实验考察了银黄颗粒在样本处理环节的回流提取溶剂的体积、回流提取时间和提取溶剂的温度等。最终选择回流提取体积为100 ml,选择回流提取时间为30 min,选择提取溶剂为热水。考察了黄芩药材在样本处理环节的回流提取溶剂的体积、回流提取时间和提取溶剂的温度等。最终选择50ml为最佳提取容积,选择40 min为最佳提取时间,选择热水提取是最佳的。考察了金银花药材在样本处理环节的回流提取溶剂的体积、回流提取时间和提取溶剂的温度等。最终选择100 ml体积提取溶剂,选择30 min提取是最佳的,选择热水提取最佳。本实验还确定了含葛根素内标的银黄颗粒、黄芩药材、金银花药材、黄芩提取物、金银花提取物供试液配制方法。[align=left][b]参考文献[/b][/align] 王亚丹,杨建波,戴忠,等.中药金银花的研究进展.药物分析杂志,2014,34(11):1928-1935 中国药典.一部.2015:1498 王彩芳,张楠,黄龙,等. HPLC法测定不同厂家银黄颗粒中黄芩苷的含量.医药论坛杂志,2006,27(24):27-28 王彩芳,黄龙,程茜,等.高效液相色谱法测定不同厂家银黄颗粒中绿原酸的含量.时珍国医国药,2007,18(5):1143-1144黄雄,黄嬛,王峻,等.银黄颗粒的HPLC特征图谱分析.药物分析杂志,2009,29(8):1320-1323 肖小河,王永炎.从热力学角度审视和研究中医药.国际生物信息与中医药论丛.新加坡:新加坡医药卫生出版社,2004:74 贺福元,罗杰英,刘文龙,等.中药谱效学研究方向方法初探.世界科学技术-中医药现代化,2004,6(6):44-50 赵渤年,于宗渊,丁晓彦,等.黄芩质量评价谱-效相关模式的研究.中草药,2011.42(2):380-383 高燕,赵渤年,于宗渊等.金银花抗流感病毒毒谱-效相关质量评价模式的研究.中华中医药杂志,2013.28(12):3508-3511 Ke Li, Wei Cheng, Xiao-Jian Liu, hu-Bin Li, En-Guang Hou, Yan Gao, Liang Wang, Qing Liu, Bo-Nian Zhao, Zong-Yuan Yu, Mathematical Modelling for the Quality Evaluation of BaikalSkullcap Root, Applied Mechanics and Materials, 2011 王荣梅,徐丽华,林永强.HPLC法同时测定银黄含片中6个咖啡酰奎宁酸类成分的含量.药物分析杂志,2012,32(1):57-60 高苏亚,范涛,王黎等.红外光谱技术结合化学计量学方法在中药研究中的应用.应用化工,2012,41(2):324-328 王鹏,王振国,薛付忠等.基于支持向量机法的中药性状与药性相关性研究. 江西中医药,2012,43(355):65-68 Cifford MN, Johnston KL, Knight S et al. Hierarchical scheme for [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url] identification ofchlorogenic acids.J Agric Food Chem,2003 51(10):2900-2910张倩,张加余,隋丞琳,等. HPLC-DAD-ESI-MS/MS研究金银花水提工艺中绿原酸类成分的变化规律.中国中药杂志,2012 37(23):3564-3567 沈红,段金廒,钱大玮,等.黄芩及复方野马追胶囊中黄酮类成分的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析.药物分析杂志,2009 29(9):1425-1429 赵胜男,李守拙.黄芩药材中黄酮类成分的HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]研究.承德医学院学报2012 29(4):345-347 Chkoshi E, Nagashima T, Sato H, et al. Simple preparation ofbaicalin from Scutellariae Rdixi. JChromatogr A,2009 1216(11): 2192 -2194高燕,吕凌,王亮,等.银黄颗粒HPLC指纹图谱与模式识别分析.中华中医药杂志,2017,32(09):4238-4242 丁晓彦,刘青,李岩,等.丹参脂溶性成分的HPLC指纹图谱及模式识别研究.中华中医药杂志,2016,3(6):2254-2256
由二甲胺与磺酰氯合成二甲胺基磺酰氯,产品计划用气相做检测,柱子是se-30的,结果不理想,大家给些意见,尤其是关于该产品的文献极少,在知网上没有几篇。也可能太简单了。
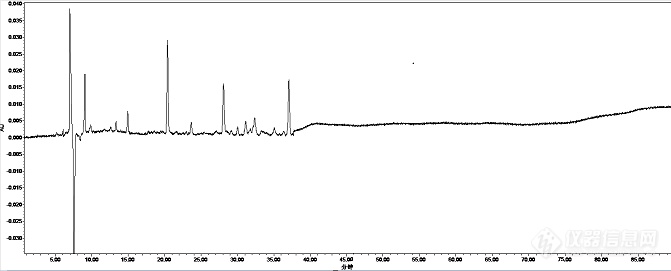
[align=center][b]一种银黄颗粒HPLC指纹图谱的检测方法及其应用[/b][/align]摘要目的:本研究介绍了银黄颗粒及其原料药黄芩和金银花的指纹图谱的建立方法,包括供试品溶液的制备、高效液相色谱仪测定及对数据和图谱的处理,以及由该方法制备得到的相应指纹图谱。方法:使用Venusil MP C18(4.6 mm × 250 mm,5 μm)+ Venusil MP C18(4.6 mm × 250 mm,3 μm)色谱柱在紫外235 nm吸收波长,选用流动相:乙腈(A)-0.3%磷酸(B)梯度洗脱,0~103 min,17%ACN 103~142 min,17%→27%ACN;142~156 min,27%→29%ACN;156~179 min,29%→41%ACN;179~219min,41%→80%CAN。结果:在测定的不同厂家 10 批次样品的色谱图中,选择90%以上批次样品均有的色谱峰为共有峰,银黄颗粒。黄芩药材、金银花药材分别确定了22,22,21 个共有峰。结论:为银黄颗粒定性鉴别提供借鉴。关键词:银黄颗粒;黄芩;金银花;指纹图谱[align=center][b]A Method for Detecting Fingerprint of Yinhuang Granules by HPLC and ItsApplication[/b][/align]Abstract Objective: To introduce the method ofestablishing fingerprint of Yinhuang granules and its raw materials,Scutellaria baicalensis and Lonicera japonica, including the preparation ofsample solution, determination by high performance liquid chromatography andthe treatment of data and chromatogram, as well as the correspondingfingerprint obtained by this method. METHODS: Venusil MP C18 (4.6 mm×250 mm, 5 mm) + Venusil MP C18 (4.6 mm×250mm, 3 mm) column was used at 235 nmultraviolet absorption wavelength. The mobile phase was selected: acetonitrile(A) - 0.3% phosphoric acid (B) gradient elution, 0-103 min, 17% ACN, 103-142min, 17% to 27% ACN, 142-156 min, 27% to 29% ACN, 156-179 min, 29% to 4% ACN.1% ACN 179 ~ 219 min, 41%80% CAN. RESULTS: In the chromatograms of 10 batchesof samples from different manufacturers, more than 90% of the samples hadcommon peaks, Yinhuang granules. Scutellaria baicalensis and Lonicera japonicahave 22, 22 and 21 peaks respectively. CONCLUSION: It can provide reference forthe qualitative identification of Yinhuang Granules.Key words: Yinhuang granules Scutellaria baicalensis Honeysuckle Fingerprint[b] 一、前言[/b]银黄颗粒组方由金银花和黄芩构成,具有清热疏风、利咽解毒的功效,用于外感风热、肺胃热盛所致的咽干、咽痛、喉核肿大、口渴、发热急慢性扁桃体炎、急慢性咽炎、上呼吸道感染等症。该复方原料金银花为忍冬科植物忍冬的干燥花蕾或带初开的花,主产于山东、河南和河北等地。该复方原料黄芩为唇形科[url=https://baike.baidu.com/item/%E9%BB%84%E8%8A%A9%E5%B1%9E][color=windowtext]黄芩属[/color][/url]多年生草本植物,产于河北,河南,陕西,山西,山东等地。黄芩提取物的主要活性成分为黄芩苷、汉黄芩苷、黄芩素及汉黄芩素,金银花提取物是从金银花中提取的有机酸类活性成分。该制剂及其原料药成分复杂,生产厂家及产地众多,样品存在差异。中药 HPLC 指纹图谱技术被认为是当前能较全面反映中药材及复方整体化学成分信息的方法,能更有效地评价中药的质量信息。本研究在分析上述研究背景的基础上,收集来源于不同产地的各50批金银花和黄芩药材,并制成银黄颗粒成品,再采用HPLC法同时建立金银花药材,黄芩药材和相应批次银黄颗粒的指纹图谱,选出各自的共有峰,从而确定不同产地,不同厂家的的药材共有物质及其数量。[b]二、材料与方法1仪器与试剂、试药1.1仪器[/b]Waters e2695高效液相色谱仪(美国Waters公司),Waters 2998紫外检测器(美国Waters公司),Waters Empower色谱工作站(美国Waters公司);AGBP210S电子天平(Sartorius公司);MILLIPORE纯水机(MILLIPORE公司);高速万能粉碎机(北京市永光明医疗仪器有限公司,FW-80型);SB4200DTS超声波双频清洗机(宁波新芝生物科技股份有限公司);KDM-A控温电热套(金坛市医疗仪器厂);Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,5 μm)和Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,3 μm)。[b]1.2 试剂与试药[/b]乙腈(上海星可高纯溶剂有限公司,色谱纯);甲醇(天津市科密欧化学试剂有限公司,色谱纯);其余试剂均为分析纯,水为超纯水。对照品来源:葛根素(批号:110752-200912)购自中国食品药品检定研究院。[b]2方法2.1 HPLC色谱条件的考察2.1.1不同流动相的考察[/b]比较了乙腈--0.05%甲酸、乙腈-0.4%甲酸、乙腈-0.3%甲酸的流动相系统进行洗脱。见图1。 [align=center][img=,671,271]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091124382201_5843_3255306_3.png!w671x271.jpg[/img][/align][align=center]乙腈--0.05%甲酸[/align][align=center][img=,610,288]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091124499241_4585_3255306_3.png!w610x288.jpg[/img][/align][align=center]乙腈-0.4%甲酸[/align][align=center][img=,690,286]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091124589941_1562_3255306_3.png!w690x286.jpg[/img][/align][align=center]乙腈-0.3%甲酸[/align][align=center][img=,610,286]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091125102591_3301_3255306_3.png!w610x286.jpg[/img][/align][align=center]图1 不同流动相系统下银黄颗粒指纹图谱[/align]结果表明,前二者分离度均相对较差,且乙腈-0.05%甲酸均基线噪音大。最终选用乙腈-0.3%磷酸作为流动相系统,所得色谱峰型较好,基线平稳,分离效果最佳。[b]2.1.2 梯度洗脱条件的选择[/b]本实验考察了不同比例的乙腈-磷酸的洗脱条件,尽可能多且全面展现银黄颗粒样品的峰信息,考察了以下4个洗脱程序。梯度条件一:流动相:乙腈(A)-0.1%磷酸(B)梯度洗脱,0~70 min,17%ACN;70~100 min,17%→20% ACN;100~110 min,20%→25% ACN;110~140 min,25%→55% ACN;140~150 min,55%→70%ACN。梯度条件二:流动相:乙腈(A)-0.1%磷酸(B)梯度洗脱,0~80min,17%ACN;80~139min,17% →34% ACN;139~159 min,34% →64% ACN;159~170min,64% →80% ACN。梯度条件三:流动相:乙腈(A)-0.1%磷酸(B)梯度洗脱,0~103min,17%ACN;103~142min,17%→24%ACN;142~165min,24%→33%ACN;165~195 min,33%ACN;195~280min,33%→70%ACN。梯度条件四:流动相:乙腈(A)-0.3%磷酸(B)梯度洗脱,0~103 min,17%ACN 103~142min,17%→27%ACN;142~156 min,27%→29%ACN;156~179 min,29%→41%ACN;179~219min,41%→80%CAN。结果梯度条件四下的指纹图谱,色谱图中采集的色谱峰形好,峰数多且分离度良好,基线较平稳,能展现最多的谱图信息,故确定为最终梯度条件,见图2。[align=center][img=,607,284]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091125509322_7749_3255306_3.png!w607x284.jpg[/img][/align][align=center]梯度条件一[/align][align=center][img=,607,287]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091126035685_7140_3255306_3.png!w607x287.jpg[/img][/align][align=center]梯度条件二[/align][align=center][img=,608,287]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091126106612_24_3255306_3.png!w608x287.jpg[/img][/align][align=center]梯度条件三[/align][align=center][img=,690,282]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091126155801_3154_3255306_3.png!w690x282.jpg[/img][/align][align=center]梯度条件四[/align][align=center]图2 不同洗脱条件下银黄颗粒指纹图谱[/align][b]2.1.3 不同流速的选择 [/b]分别考察同一样品供试液,以梯度条件四下的方法,测定其流速在0.9 mLmin[sup]-1[/sup]、 0.8 mLmin[sup]-1[/sup]、 0.7mLmin[sup]-1[/sup]时的分离效果。结果表明,流速在0.9 mLmin[sup]-1[/sup]和0.8mLmin[sup]-1[/sup]时,130min附近两峰分离效果不理想,而0.7 mLmin[sup]-1[/sup]时峰形及分离情况均比较理想。综合考虑,选择0.7 mLmin[sup]-1[/sup]流速。见图3。[align=center][img=,690,283]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091126346021_2490_3255306_3.png!w690x283.jpg[/img][/align][align=center]流速:0.9 mLmin[sup]-1[/sup][/align][align=center][sup][img=,690,264]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091126456661_6012_3255306_3.png!w690x264.jpg[/img][/sup][/align][align=center]流速:0.8 mLmin[sup]-1[/sup][/align][align=center][sup][img=,690,286]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091127029111_5432_3255306_3.png!w690x286.jpg[/img][/sup][/align][align=center]流速:0.7 mLmin[sup]-1[/sup][/align][align=center]图3 不同流速下银黄颗粒指纹图谱[/align][b]2.1.4 测定波长的选择 [/b]对同一银黄颗粒样品供试液在235~295 nm波长范围内,每隔20 nm测定一次,选择最佳吸收波长。其色谱图结果见图4。[align=center][img=,690,285]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091127570951_5526_3255306_3.png!w690x285.jpg[/img][/align][align=center]235 nm[/align][align=center][img=,690,283]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091128103641_8767_3255306_3.png!w690x283.jpg[/img][/align][align=center]255 nm [/align][align=center][img=,690,284]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091128182679_3036_3255306_3.png!w690x284.jpg[/img][/align][align=center]275 nm[/align][align=center][img=,690,236]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091128256647_2872_3255306_3.png!w690x236.jpg[/img][/align][align=center]295 nm[/align][align=center] 图4 不同波长下银黄颗粒指纹图谱[/align]由图4结果可知,在235 nm时,色谱图中峰形佳,各峰间比例协调,基线较平稳,且呈现的峰信息量大。因此,选用235 nm作为测定波长。[b]2.2 不同色谱柱的考察[/b]考虑到银黄颗粒中主要是黄酮类成分,故选用C[sub]18[/sub]柱,对色谱柱进行考察,分别使用VenusilMP C[sub]18[/sub](4.6 mm × 250 mm,5 μm),Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,3 μm),Agela MP S/N.三根色谱柱及其不同组合,在同一梯度条件下分别对同一银黄颗粒供试液进行指纹图谱峰的采集,结果前四根色谱柱分离度相对较差,Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,5 μm)+ Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,3 μm)组合柱分离出的色谱峰较多,峰型较好,对流动相条件进行微调后,进行色谱图的采集。见图5。[align=center][img=,690,236]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091131336001_8004_3255306_3.png!w690x236.jpg[/img][/align][align=center]Agela MP S/N(4.6 mm × 250 mm,3 μm)[/align][align=center][img=,690,236]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091131265708_7298_3255306_3.png!w690x236.jpg[/img][/align][align=center]Agela MP S/N(4.6 mm × 250 mm,3 μm) [/align][align=center][img=,690,233]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091131195130_7680_3255306_3.png!w690x233.jpg[/img][/align][align=center]Venusil MP C[sub]18[/sub](4.6 mm× 250 mm,3 μm) [/align][align=center][img=,690,233]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091131092861_3850_3255306_3.png!w690x233.jpg[/img][/align][align=center]Agela MP S/N(4.6 mm × 250 mm,3 μm)+Venusil MP C[sub]18[/sub](4.6 mm× 250 mm,5 μm) [/align][align=center] [img=,690,285]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091130395070_4225_3255306_3.png!w690x285.jpg[/img][/align][align=center]Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,5 μm)+ Venusil MP C[sub]18[/sub](4.6 mm × 250 mm,3 μm)[/align]图5 不同色谱柱银黄颗粒指纹图谱[b]2.3 方法学考察2.3.1 仪器精密度考察 [/b]取银黄颗粒同一样品供试液,10 μL进样,连续进样 5次,按“4.1”项下的色谱条件进样测定,以葛根素为参照峰,计算共有峰的峰面积和相对保留时间比值。结果显示各共有峰的相对峰面积RSD<3 %,相对保留时间RSD<3%,表明仪器精密度良好。见表1、2。[align=center]表1 银黄颗粒指纹图谱精密度(相对峰面积)[/align][align=center][img=,348,494]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091132223573_3518_3255306_3.png!w348x494.jpg[/img] [/align][align=center]表2 银黄颗粒指纹图谱精密度(相对保留时间)[/align][align=center][img=,352,511]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091132363167_2471_3255306_3.png!w352x511.jpg[/img] [/align][b]2.3.2 重复性试验 2.3.2.1银黄颗粒重复性实验考察[/b]银黄颗粒研细后精密称取细粉1.0 g,称取五份分别置于100 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水100 ml于圆底烧瓶中,称重,加热回流30 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算相对峰面积值和相对保留时间值,结果见表3、4的25个峰的RSD值都接近3%,由此可以得出结论,银黄颗粒的重复性良好。[align=center]表3 银黄颗粒指纹图谱重复性(相对峰面积)[/align][align=center][img=,294,424]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091133158091_6693_3255306_3.png!w294x424.jpg[/img][/align][align=center] 表4 银黄颗粒指纹图谱重复性(相对保留时间)[/align][align=center][img=,301,407]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091133264387_6060_3255306_3.png!w301x407.jpg[/img][/align][b]2.3.2.2黄芩药材重复性实验考察[/b]黄芩药材研细后精密称取细粉0.57 g,称取五份分别置于100 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水50 ml于圆底烧瓶中,称重,加热回流40 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算相对峰面积值和相对保留时间值,结果见表5、6的15个峰的RSD值都接近3 %,由此可以得出结论,黄芩药材的重复性良好。[align=center][b] [/b]表5黄芩药材指纹图谱重复性(相对峰面积)[/align][align=center][img=,521,450]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091134133924_7231_3255306_3.png!w521x450.jpg[/img][/align][align=center]表6 黄芩药材指纹图谱重复性(相对保留时间)[/align][align=center][img=,526,450]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091134223683_8185_3255306_3.png!w526x450.jpg[/img][/align][b]2.3.2.3金银花药材重复性实验考察[/b]金银花药材研细后精密称取细粉0.5 g,称取五份分别置于100 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水100 ml于圆底烧瓶中,称重,加热回流30 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算相对峰面积值和相对保留时间值,结果见表7、8的15个峰的RSD值都接近3%,由此可以得出结论,金银花药材的重复性良好。[align=center] 表7 金银花药材指纹图谱重复性(相对峰面积)[/align][align=center][img=,521,453]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091135202061_4447_3255306_3.png!w521x453.jpg[/img][/align][align=center]表8 金银花药材指纹图谱重复性(相对保留时间)[/align][align=center][img=,520,450]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091135276281_796_3255306_3.png!w520x450.jpg[/img][/align][b]2.4.3 稳定性试验 2.4.3.1银黄颗粒稳定性实验考察[/b]银黄颗粒研细后精密称取细粉1.0 g置于100 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水100 ml于圆底烧瓶中,称重,加热回流30 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算相对峰面积值和相对保留时间值,结果见表9、10的25个峰的RSD值都接近3 %,由此可以得出结论,银黄颗粒的稳定性良好。[align=center]表9 银黄颗粒指纹图谱稳定性(相对峰面积)[/align][align=center][img=,284,411]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091135513432_6898_3255306_3.png!w284x411.jpg[/img] [/align][align=center]表10 银黄颗粒指纹图谱稳定性(相对保留时间)[/align][align=center][img=,285,423]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091135598981_6279_3255306_3.png!w285x423.jpg[/img][/align] [b]2.4.3.2黄芩药材稳定性实验考察[/b]黄芩药材研细后精密称取细粉0.57 g置于50 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水100 ml于圆底烧瓶中,称重,加热回流40 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算相对峰面积值和相对保留时间值,结果见表11、12的25个峰的RSD值都接近3 %,由此可以得出结论,黄芩药材的稳定性良好。[align=center] 表11 黄芩药材指纹图谱稳定性(相对峰面积)[/align][align=center][img=,534,451]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091136295090_8710_3255306_3.png!w534x451.jpg[/img][/align][align=center] 表12 黄芩药材指纹图谱稳定性(相对保留时间)[/align][align=center][img=,468,403]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091136437515_7524_3255306_3.png!w468x403.jpg[/img][/align][b]2.4.3.3金银花药材稳定性实验考察[/b]金银花药材研细后精密称取细粉1.0 g置于100 ml 的圆底烧瓶中,精密加入煮沸的蒸馏水100 ml于圆底烧瓶中,称重,加热回流30 min,回流后放冷,补重,过滤,取续滤液。将样品溶液与内标溶液经0.45 μm微孔滤膜滤过后等体积混匀,作为供试品溶液,注入高效液相色谱仪,按照既定方法采集色谱指纹图谱,计算相对峰面积值和相对保留时间值,结果见表13、表14,25个峰的RSD值都接近3 %,由此可以得出结论,金银花药材的稳定性良好。[align=center]表13 金银花药材指纹图谱稳定性(相对峰面积)[/align][align=center][img=,452,406]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091137142831_7122_3255306_3.png!w452x406.jpg[/img][/align][align=center]表14 金银花药材指纹图谱稳定性(相对保留时间)[/align][align=center][img=,441,402]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091137227371_5231_3255306_3.png!w441x402.jpg[/img][/align][b]2.5 样品共有峰的确定[/b]对10 批不同厂家的银黄颗粒及黄芩和金银花药材供试液进行分析,采集指纹图谱,并以葛根素作为参考峰,银黄颗粒选取标定了22个共有峰,见图6。黄芩药材选取标定了22个共有峰,见图7。金银花药材选取标定了21个共有峰,见图8。[align=center][img=,690,265]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091137498881_3854_3255306_3.png!w690x265.jpg[/img][/align][align=center]图6 10 批次的银黄颗粒共有特征峰[/align][align=center][img=,690,259]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091138017531_1112_3255306_3.png!w690x259.jpg[/img][/align][align=center]图7 10 批次的黄芩共有特征峰[/align][align=center][img=,690,247]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091138139735_5366_3255306_3.png!w690x247.jpg[/img][/align][align=center]图8 10 批次金银花药材的共有特征峰[/align][b]三、结果与分析[/b]本研究介绍了银黄颗粒及其原料药黄芩和金银花的指纹图谱的建立方法,包括供试品溶液的制备、高效液相色谱仪测定及对数据和图谱的处理,以及由该方法制备得到的相应指纹图谱。在测定的不同厂家 10 批次样品的色谱图中,选择90 %以上批次样品均有的色谱峰为共有峰,银黄颗粒。黄芩药材、金银花药材分别确定了22,22,21 个共有峰。[b]四、讨论与结论[/b]本研究的指纹图谱构建方法操作简单,稳定可靠,精密度高,分离度好,指纹图谱的稳定性和重现性较好,且信息量大,采用指纹图谱找出不同产地,不同厂家的同一药材的共有峰为质量控制手段,既避免了因只测定一、两个化学成分而判定制剂整体质量的片面性,又减少了为质量达标而人为处理的可能性,通过对多个批次的样品进行系统分析,能更加全面、科学评价银黄颗粒的质量,从而使产品的质量和疗效得到保证。参考文献王亚丹,杨建波,戴忠,等.中药金银花的研究进展.药物分析杂志,2014,34( 11):1928-1935 中国药典.一部.2015:1498 王彩芳,张楠,黄龙,等. HPLC法测定不同厂家银黄颗粒中黄芩苷的含量.医药论坛杂志,2006,27 (24):27-28 王彩芳,黄龙,程茜,等.高效液相色谱法测定不同厂家银黄颗粒中绿原酸的含量.时珍国医国药,2007,18(5):1143-1144黄雄,黄嬛,王峻,等.银黄颗粒的HPLC特征图谱分析.药物分析杂志,2009,29(8):1320-1323 肖小河,王永炎.从热力学角度审视和研究中医药.国际生物信息与中医药论丛.新加坡:新加坡医药卫生出版社,2004:74 贺福元,罗杰英,刘文龙,等.中药谱效学研究方向方法初探.世界科学技术-中医药现代化,2004,6(6):44-50 赵渤年,于宗渊,丁晓彦,等.黄芩质量评价谱-效相关模式的研究.中草药,2011.42(2):380-383 高燕,赵渤年,于宗渊等.金银花抗流感病毒毒谱-效相关质量评价模式的研究.中华中医药杂志,2013.28(12):3508-3511 Ke Li, Wei Cheng, Xiao-Jian Liu,hu-Bin Li, En-Guang Hou, Yan Gao, Liang Wang, Qing Liu, Bo-Nian Zhao, Zong-Yuan Yu, Mathematical Modelling for the Quality Evaluation of Baikal Skullcap Root, Applied Mechanics and Materials, 2011 王荣梅,徐丽华,林永强.HPLC法同时测定银黄含片中6个咖啡酰奎宁酸类成分的含量.药物分析杂志,2012,32(1):57-60 高苏亚,范涛,王黎等.红外光谱技术结合化学计量学方法在中药研究中的应用.应用化工,2012,41(2):324-328 王鹏,王振国,薛付忠等.基于支持向量机法的中药性状与药性相关性研究. 江西中医药,2012,43(355):65-68 Cifford MN, Johnston KL, Knight S et al. Hierarchical scheme for [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url] identification of chlorogenic acids.J Agric Food Chem,2003 51(10):2900-2910张倩,张加余,隋丞琳,等. HPLC-DAD-ESI-MS/MS研究金银花水提工艺中绿原酸类成分的变化规律.中国中药杂志,2012 37(23):3564-3567 沈红,段金廒,钱大玮,等.黄芩及复方野马追胶囊中黄酮类成分的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析.药物分析杂志,2009 29(9):1425-1429 赵胜男,李守拙.黄芩药材中黄酮类成分的HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]研究.承德医学院学报2012 29(4):345-347 Chkoshi E, Nagashima T, Sato H, et al. Simple preparation of baicalin from Scutellariae Rdixi. J Chromatogr A,2009 1216(11): 2192 -2194高燕,吕凌,王亮,等.银黄颗粒HPLC指纹图谱与模式识别分析.中华中医药杂志,2017,32(09):4238-4242 丁晓彦,刘青,李岩,等.丹参脂溶性成分的HPLC指纹图谱及模式识别研究.中华中医药杂志,2016,3(6):2254-2256
二甲胺基磺酰氯在合成过程中在主峰前出现一个1%的左右的杂质,请教高手帮我解疑是什么?
HPLC法测小儿氨酚黄那敏颗粒中马来酸氯苯那敏的含量。本人查阅了一些文献,各篇文章中均不一样。①大连依利特C18(250mm*4.6mm,5um);流动相是甲醇-0.5mol/l磷酸二氢钠-三乙胺(150:850:0.25),用磷酸调节pH至2.3;检测波长为215nm②Eclipse*DB-C18(250mm*4.6mm,5um);流动相是甲醇-0.05mol/l磷酸二氢钾-三乙胺(10:90:0.02),用磷酸调节pH至3.4;检测波长为215nm③Shim-pack VP-ODS(150mm*4.6mm,5um);流动相是甲醇-0.05mol/l磷酸二氢钠-三乙胺(19:81:0.025),用磷酸调节pH至2.2;检测波长为215nm④Kromasil C18(250mm*4.6mm,5um);流动相是乙腈-0.3%十二烷基硫酸钠溶液-磷酸(60:40:0.02),用三乙胺调pH至3.5;检测波长为254nm有没有谁能帮我分析一下?我们现在有大连依利特和美国天地的柱子,岛津液相色谱仪。谢谢!!!
磺胺药物对氨基苯磺酰胺的合成目的原理Ar-NHCOCH3 + 2HOSO2Cl → p-ClO2S-Ar-NHCOCH3+ HClp-ClO2S-Ar-NHCOCH3 + NH3 → p-CH3CONH-Ar-SO2NH2 + HClp-CH3CONH-Ar-SO2NH2 + H2O → p-H2N-Ar-SO2NH2 + CH2CO2H仪器药品乙酰苯胺(自制) 5g(0.037mol);氯磺酸(d=1.77) 22.5g(12.5ml,0.19mol);浓氨水(28%,d=0.9) 35ml 浓盐酸,碳酸钠。过程步骤(1)对乙酰氨基苯碘酰氯在100ml干燥的锥形瓶中,加入5g干燥的乙酰苯胺,在石棉网上用小火加热熔化。瓶壁上若有少量水气凝结,应用干净的滤纸吸去。冷却使熔化物凝结成块。将锥形瓶置于冰浴中冷却后,迅速倒入12.5ml氯磺酸,立即塞上带有氯化氢导气管的塞子。反应很快发生,若反应过于激烈,可用冰水浴冷却。待反应缓和后,旋摇锥形瓶使固体全溶,然后再在温水浴中加热10~15min使反应完全。将反应瓶在冷水中充分冷却后,于通风中在充分搅拌下,将反应液慢慢倒入盛75g碎冰的烧杯,用少量冷水洗涤反应瓶,洗涤液倒入烧杯中。搅拌数分钟,并尽量将大块固体粉碎,使成颗粒小而均匀的白色固体。抽滤收集,用少量冷水洗涤,压干,立即进行下一步反应。(2)对乙酰氨基苯磺酰胺将上述粗产物移入烧杯中,在不断搅拌中慢慢加入17.5ml浓氨水(在通风橱内),立即发生放热反应并产生白色糊状物。加完后,继续搅拌15min,使反应完全。然后加入19ml水,在石棉网上用小火加热10~15min,并不断搅拌,以除去多余的氨,得到的混合物可直接用于下一步合成。(3)对氨基苯磺酰胺(磺胺)将上述反应物放入圆底烧瓶中,加入3.5ml浓盐酸,在石棉网上用小火加热回流0.5h。冷却后,应得一几乎澄清的溶液,若有固体析出,应继续加热,使反应完全。如溶液呈黄色,并有极少量固体存在时,需加入少量活性炭煮沸10min,过滤。将滤液转入大烧杯中,在搅拌下小心加入粉状碳酸钠至恰呈碱性(约4g)。在冰水浴中冷却,抽滤收集固体,用少量冰水洗涤,压干。粗产物用水重结晶(每克产物约须12ml水),产量3~4g。熔点161~162℃。纯品对氨基苯磺酰胺为白色针状结晶,熔点163~164℃。注意事项1.氯磺酸对皮肤和衣服有强烈的腐蚀性,暴露在空气中会冒出大量氯化氢气体,遇水会发生猛烈的放热反应,甚至爆炸,故取用时需加小心。反应中所用仪器及药品皆需十分干燥,含有氯磺酸的废液不可倒入水槽,而应倒入废液缸中。工业氯磺酸常呈棕黑色,使用前宜用磨口仪器蒸馏纯化,收集148~150℃的馏分。2.酰磺酸于乙酰苯胺的反应非常剧烈,将乙酰苯胺凝结成快状,可使反应缓和进行,当反应过于激烈时,应适当冷却。3.在氯磺化过程中,将有大量氯化氢气体放出。为避免污染室内空气,装置应严密,导气管的末端要与接受器内的水面接近,但不能插入水中,否则可能倒吸而引严重事故!4.加入速度必须缓慢,必须充分搅拌,以免局部过热而使对乙酰胺基苯磺酰胺水解。这是实验成功的关键。5.尽量洗去固体所夹杂和吸附的盐酸,否则产物在酸性介质中放置过久,会很快水解,因此在洗涤后,应尽量压干,且在1~2h内将它转变为磺胺类化合物。6.粗制的对氨基苯磺酰氯久置容易分解,甚至干燥后也不可避免。若要得到纯品,可将粗产物溶于温热的氯仿中,然后迅速转移到事先温热的分液漏斗中,分出氯仿层,在冰水浴中冷却后即可析出晶体。纯品对氨基苯磺酰氯的熔点为149℃。7.为了节省时间,这一步的粗产物可不必分出。若要得到产品,可在冰水浴中冷却,抽滤,用冰水洗涤,干燥即可。粗品用水重结晶,纯品熔点为219~220℃。8.对乙酰胺基苯磺酰胺在稀酸中水解成磺胺,后者又与过量的盐酸形成水溶性的盐酸盐,所以水解完成后,反应液冷却时应无晶体析出。由于水解前溶液中氨的含量不同,加3.5ml盐酸有时不够,因此,在回流至固体全部消失前,应测一下溶液的酸碱性,若酸性不够,应补加盐酸回流一段时间。9.用碳酸钠中和滤液中的盐酸时,有二氧化碳产生,故应控制加热速度并不断搅拌使其逸出。磺胺是一两性化合物,在过量的碱溶液中也易变成盐类而溶解。故中和操作必须仔细进行,以免降低产量。分析思考 1.为什么在氯磺化反应完成以后处理反应混合物时,必须移到通风橱中,且在充分搅拌下缓缓倒入碎冰中?若在未倒完前冰就化完了,是否应补加冰块?为什么?2.为什么苯胺要乙酰化后在氯磺化?直接氯磺化行吗?3 .如何理解对氨基苯磺酰氨是两性物质?试用反应式表示磺胺与稀酸和稀碱的作用。
当固定污染源颗粒物低于检出限,那排放速率是要按检出限的1/2进行计算吗,看到好多检测报告都是这么算的,这个计算方法是在哪个标准里有提到呢?
我做生物胺的柱前衍生,用的是10mg/ml的丹磺酰氯丙酮容液,但是配完之后丹磺酰氯很多都沉在底部并不溶解,用超声混匀后,用移液枪吸取能明显看到颗粒。然后将丹磺酰氯丙酮容液加进生物胺单标里混匀后会底部出现白色沉淀,是不是有发生什么化学反应?这个沉淀看了很多文献都没提到过。然后就是去跑液相,8个标品,只有精氨酸一直跑不出,时间感觉也很充足了,就是一直很平坦不出峰,是不是和上面的操作有关系啊?
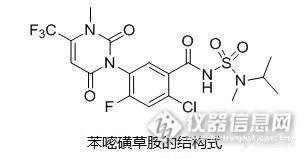
上世纪60年代。当时,杜邦公司开发出了首个脲嘧啶类除草剂—除草定,正式开启了该类除草剂研发的先河。而真正掀起脲嘧啶类除草剂开发热潮的是在上世纪90年代,当时人们对于该类除草剂的作用机理有了更深入的了解,发现脲嘧啶类除草剂属于原卟啉原氧化酶(PPO)抑制剂。杜邦公司在推出除草定后,又相继推出了异草定和特草定等产品。富美实的双苯嘧草酮以及先正达的氟丙嘧草酯均属于该类除草剂。而巴斯夫于2009年推出的苯嘧磺草胺(saflufenacil)更属于该类除草剂中的佼佼者。苯嘧磺草胺能够适用于多种生产系统和非耕地,在苗后或苗前均能使用;其次,适用作物多。苯嘧磺草胺能够用于包括谷物、玉米、棉花、水稻、高粱、大豆和果树等在内的30多种作物上;再次,防除谱广。苯嘧磺草胺能够防除90余种阔叶杂草,包括一些对三嗪类、草甘膦及乙酰乳酸合成酶抑制剂存在抗性的杂草。另外,它也具有作用快、残效期长等多种特性。http://ng1.17img.cn/bbsfiles/images/2017/02/201702010042_01_1623180_3.jpg2009年,苯嘧磺草胺在南美国家尼加拉瓜、智利和阿根廷三国登记。2010年,苯嘧磺草胺与精二甲吩草胺的复配制剂Verdict在美国获得登记,用于大豆。同年,苯嘧磺草胺正式登陆中国,以70%水分散粒剂(商品名:巴佰金)的形式面世,用于柑橘园和非耕地的杂草防除,由诺普信负责在中国市场的总经销。目前,苯嘧磺草胺已在美国、加拿大、中国、尼加拉瓜、智利、阿根廷、巴西和澳大利亚等国登记。苯嘧磺草胺可替代苯氧类除草剂2,4-D和磺酰脲类除草剂与草甘膦复配,可降低防治顽固性杂草对草甘膦的使用量。2014年,苯嘧磺草胺的全球销售额达到1.4亿美元。据巴斯夫公司预测,苯嘧磺草胺可实现3亿欧元的年峰值销售额。苯嘧磺草胺目前仍处于专利保护期中,其在中国的专利为巴斯夫于2001年申请的《尿嘧啶取代的苯基氨磺酰羧酰胺》,专利号为ZL01801896.3,对苯嘧磺草胺的化合物及合成方法进行了保护,该专利将于2021年4月30日到期.

今天在药店见到好多小儿氨酚黄那敏颗粒(检测标准见40楼),包装精美图案精致,各个厂家对包装上是下了不少功夫了?土豆想:一个品种这么多药厂生产,大家都符合药典规定,质量上无法分出高低,只好在别的方面一较高下了。最近搜集好妹妹上瘾了,看到不同的小儿氨酚黄那敏颗粒我就拍下来,也向各位征集图片了,凡是能提供和我这不重复的小儿氨酚黄那敏颗粒图片的,一张图加10分,十分啊!!http://ng1.17img.cn/bbsfiles/images/2011/08/201108251407_312180_1645752_3.jpg好可爱的小宝宝啊,是不是?http://ng1.17img.cn/bbsfiles/images/2011/08/201108251407_312182_1645752_3.jpg这是个小鸡还是小龙人的卡通图案呢?http://ng1.17img.cn/bbsfiles/images/2011/08/201108251408_312183_1645752_3.jpg宝宝看到这么可爱的图案,一定减少了吃药的抗拒性了。http://ng1.17img.cn/bbsfiles/images/2011/08/201108251409_312184_1645752_3.jpg这个卡通好像不太可爱了http://ng1.17img.cn/bbsfiles/images/2011/08/201108251410_312186_1645752_3.jpg佩夫人登场-----
2011年3月14日,加拿大卫生部拟修订啶酰菌胺、赛座灭、甲磺草胺和甲基环丙烯等4种物质的最大残留限量标准。将啶酰菌胺在番杏和菠菜中的最大限量标准修订为60 mg/kg,刺菜蓟、芹菜、香芹、莴笋、大黄、唐莴苣、茴香叶(茎)中的最大限量标准修订为45mg/kg;将甲磺草胺在干制大豆中的最大限量标准修订为0.05 mg/kg,薄荷中的最大残留限量标准修订为0.3mg/kg,大白菜,山葵根和葵花籽中的最大残留限量标准修订为0.15 mg/kg,干制去壳豌豆中的最大残留限量标准修订为0.15mg/kg;将甲基环丙烯在梨中的最大残留限量标准修订为0.01mg/kg;将赛座灭在牛肉、山羊肉、绵羊肉、马肉及其副产品和牛奶等产品中的最大残留限量标准修订为0.02 mg/kg.
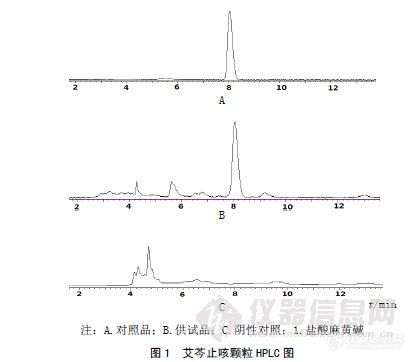
作者:王朋义; 张海鸣; 王成芳; 曹杰; 杜树山;(内蒙古伊泰药业有限责任公司; 北京师范大学资源学院教育部资源药物工程研究中心;)摘要:目的建立艾芩止咳颗粒中盐酸麻黄碱的高效液相含量测定方法。方法采用Diamonsil C18(4.6 mm×250 mm,5μm)色谱柱,以乙腈-0.025 mol/L磷酸溶液(10∶90)为流动相,流速为1 mL/min,检测波长为210 nm,柱温30℃。结果盐酸麻黄碱含量测定的线性范围为0.13~0.77μg(r=0.99992),精密度试验RSD=0.11%。平均加样回收率为97.87%,RSD=1.68%。结论本方法快速、简便、准确、重复性较好,结果可靠,可为艾芩止咳颗粒的质量评价提供有效手段。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061422_381882_1606903_3.jpg
二甲胺基磺酰氯在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]中出峰吗?
大家好,请问“颗粒”点用什么仪器设备能准确分析元素成分
请问专家,我做颗粒度时设置仪器自动分析三次,是什么原因导致了三次的粒径范围相差很大,而每次的平均粒径却差不多?我用的是贝克曼的N5颗粒度计,测试角为90度,样品为稀释过的丁苯胶乳
目前关于固定污染源废气 颗粒物的测定,我们只能做GB/T 16157,那么在接项目的时候,如果涉及不确定颗粒物浓度高低的情况下,我们可以用GB/T 16157去做吗?如果结果低于20mg/m3,就报低于检出限? 还需要用HJ 836的标准去定量分析吗?目前我们没有HJ 836的能力。除了扩项之外,面对这种情况有什么解决方案吗?
现在先进的激光颗粒测试仪(激光颗粒分析仪) 不管国内国外的能测试颗粒粒径 最小 10nm 左右的麻烦清楚的告诉下 型号 规格 厂家 最好有联系方式
奢华的晚餐,上好的美酒,总令人向往,而数小时后,快速心跳和剧烈头痛总是接踵而至,难以避免。但是伯克利加州大学的学者们最新推出的设备似乎可以避免这令人恐惧的”酒后头痛”。 化学家们和国家航空和宇宙航行局一起研究寻找火星上生物的技术,开发了一个新设备。据他们的描述,这个设备轻易探测到了一些化学物质,许多化学家们都认为这些物质是使葡萄酒和其他嗜好性食物变成令人身体不舒服的物质。 这种化学物质,叫做生物胺,它广泛的存在于被美食家大家赞誉的腌制食品、发酵食品和陈年的食品中。这些食物包括葡萄酒、巧克力、奶酪、橄榄、坚果和深加工的肉制品。 在分析化学期刊发表这项新技术文章的作者Richard Mathies,说“你想象不出你吃的食物与你体内的化学物质的关系有多么紧密。”
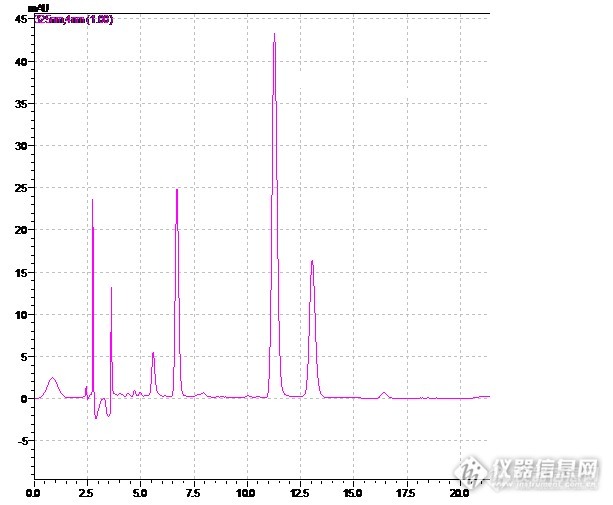
摘要: 目的 建立茵栀黄颗粒中栀子苷和绿原酸的含量测定方法。方法 采用高效液相色谱法。色谱柱:Kromasil C18柱(4.6mm×250mm,5μm);流动相:乙腈-0.1%甲酸(10:90);流速1.0ml/min;柱温:35℃;检测波长:238nm、325nm。结果:对茵栀黄颗粒中栀子苷和绿原酸进行含量测定。 栀子苷在10.43~52.15μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=123721821.7x-6951.8(r=0.9999)。平均加样回收率为97.21% RSD为1.62% (n=6) 绿原酸在10.74~53.70μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=29447356x-25436(r=0.9999)。平均加样回收率为96.61% RSD为1.35% (n=6)结论 该方法操作简便,结果可靠,可用于茵栀黄颗粒中栀子苷和绿原酸的含量测定。 清茵栀黄颗粒,清热解毒,利湿退黄。有退黄疸和降低谷丙转氨酶的作用。用于湿热毒邪内蕴所致急性、慢性肝炎和重症肝炎(I型)。也可用于其他型重症肝炎的综合治疗。由陈提取物、栀子提取物、黄芩苷、金银花提取物组成。为了进一步控制药品质量,我们对其中的进行了栀子苷和绿原酸含量测定,以期为后续进一步研究奠定基础!关键词:HPLC;DAD;茵栀黄颗粒;栀子苷 ;绿原酸1 仪器与试药1.1 仪器 岛津LC-20AT高效液相色谱仪,SPD-M20A检测器, LCsolution色谱工作站, Metteler AB265S(0.01mg);Sartorius BS124S(0.1mg)),必能信超声波清洗器(必能信超声有限公司)1.2 试药 绿原酸对照品(批号110753-201314)、栀子苷对照品(批号:110749-201115),均购自中国食品药品检定研究院;乙腈(色谱纯);其它试剂均为国产分析纯,蒸馏水自制。2 实验方法与结果2.1 色谱条件与图谱 色谱柱: Kromasil C18柱(4.6mmx250mm,5um); 检测波长:238nm;325nm 流速:1.0ml.min-1; 流动相:乙腈-0.1%甲酸(10:90); 柱温:35℃。2.2 溶液制备2.2.1对照品溶液 取栀子苷和绿原酸对照品适量,精密称定,加甲醇制成每1ml 分别含0.1043mg和0.1074mg 的混合溶液,即得。2.2.2供试品溶液 取本品,研细,取约0.5g,精密称定置50ml量瓶中,精密加入甲醇40ml,称定重量,超声处理30min ,再称定重量,用甲醇补足减失的重量,定容至50ml,摇匀,滤过,即得。2.3 线性关系考察 精密量取各对照品溶液,制得5个系列不同浓度的对照品溶液,各取10微升依次进样,按上述色谱条件测定峰面积,以对照品峰面积Y为纵坐标,浓度X(mg·mL )为横坐标,绘制标准曲线,结果栀子苷在10.43~52.15μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=123721821.7x-6951.8(r=0.9999)。绿原酸在10.74~53.70μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=58134683.4x-7988.3(r=0.9999) http://ng1.17img.cn/bbsfiles/images/2015/10/201510020006_569010_1839779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015100200003568_01_1839779_3.png2.4精密度试验 精密吸取同一供试品溶液,重复进样6次,计算栀子苷和绿原酸对照品 峰面积的RSD分别为0.4% 和1.4% 。2.5 稳定性试验 精密吸取同一供试品溶液,分别于0,2,4,8,16,24 h测定,结果表明:栀子苷和绿原酸对照品在24 h内稳定,RSD分别为1.1% 和1.5% 。2.6 重复性实验 取同一批次的茵栀黄颗粒平行6份,制成供试品溶液,在上述色谱条件下分别进行HPLC分析,计算栀子苷和绿原酸对照品峰面积的RSD分别为1.2%和1.4% 。2.7 回收试验 取同一批茵栀黄颗粒,约取0.25g,共计6份,精密称定,按上述色谱条件进样测定,用回归方程计算回收率。2.8 样品含量测定 取3个批号的茵栀黄颗粒,配制成供试品溶液,在上述色谱条件下进行测定,栀子苷和绿原酸对照品的含量,结果见表。样品编号栀子苷(mg/g)20150729 2.54201410032.61201502162.65样品编号绿原酸(mg/g)20150729 1.04201410031.16201502161.12http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111035245_01_1839779_3.bmp 图1 茵栀黄颗粒的HPLC图谱(238nm)http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111125995_01_1839779_3.bmp 图2 栀子苷的HPLC图谱(238nm)http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111105517_01_1839779_3.png 图3 茵栀黄颗粒的HPLC图谱(325nm)http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111150072_01_1839779_3.bmp 图4 绿原酸的HPLC图谱(325
目录 一.概述 二.验证目的 三.验证小组组成 四.清洗方法及检测方法 五.取样方法检测方法及残留量的计算 六.检测结果 七.偏差及变更的处理 八.验证结论 九.再验证 十.验证结果 一. 概述 204车间无菌精烘包内生产的是环磷酰胺,其设备均为专用设备,为了防止批与批之间的污染,特制定204车间无菌精烘包摇摆式颗粒机的清洗验证报告。 二.验证目的 1) 为摇摆式颗粒机清洗程序的建立提供依据。 2) 证明该清洗方法充分有效。 3) 摇摆式颗粒机清洗后,残留物符合生产下一批要求。 三.验证小组组成 组成 部门/姓名 工作内容 组长: 验证报告批准 组员: 验证报告审核 组员: 验证报告起草 组员: 验证实施过程中的监督,管理 四.清洗及检测方法 清洗方法:将摇摆式颗粒机的过筛部分取下,送至洗涤间,用注射用水反复冲洗,直至目视无残留。再用注射用水冲洗1分钟,倒扣在架子上晾3分钟。 检测方法:以注射用水润湿棉球擦拭摇摆式颗粒机加料斗面积25 cm2。擦拭棉球以注射用水分三次萃取,萃取液以注射用水稀至50ml。 进行外观检查和HPLC检查。
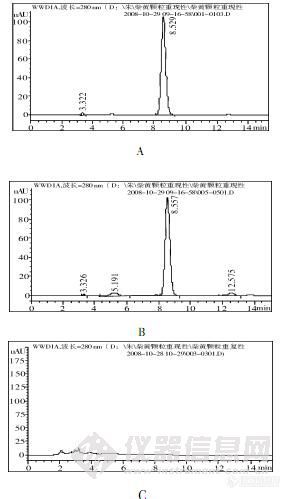
作者:黄妙婵(东莞市人民医院 药剂科, 广东 东莞 523018)摘要: 目的建立柴黄颗粒中黄芩苷的含量测定方法。方法采用迪马钻石C18(4.6mm×250mm,5μm)为色谱柱,以甲醇-0.4%磷酸(55∶45)为流动相,检测波长为280nm。结果黄芩苷在0.131μg~1.309μg范围内呈良好的线性关系,r=1.000,平均加样回收率为100.95%,相对标准偏差值(RSD)=1.38%。结论本法操作简便,结果准确、可靠、重现性好,可用于柴黄颗粒的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/07/201207161426_377871_1606903_3.jpg

7月22至26日,国家环境分析测试中心主任黄业茹一行来到天瑞仪器,就环保部《空气和废气中颗粒物测定-XRF法标准》开题验证事项进行专项考察与实验。 《空气和废气中颗粒物测定-XRF法标准》由国家环境分析测试技术研究室李玉武主任负责撰写。国家环境分析测试中心主任黄业茹、副主任吴忠祥、分析测试技术研究室主任李玉武、苏州环境监测中心站副站长顾钧、分析室主任顾海东等领导,与天瑞仪器董事长刘召贵博士、总经理应刚、副总经经理胡晓斌、应用研发中心主任姚栋梁博士、研发一部副部长吴升海博士,共同对该标准制定进行了深入探讨。天瑞仪器作为验证单位参与标准的制定工作,并组织现场验证。同期,双方还表达了拓宽合作、共建实验室的意愿。 刘召贵博士表示,国家环境分析测试中心从事着最顶尖的环境科学分析测试技术和方法研究,承担过多项国家科技攻关课题、重大工程项目。天瑞非常荣幸能够参与标准的制定工作。“并希望能充分发挥天瑞在环境监测方法、技术和仪器研发、制造、服务等方面的优势,从而与国家环境分析测试中心在环境监测仪器应用技术、经验、资源等方面实现优势互补。” 黄业茹主任在洽谈中强调,近年来,国产仪器发展势头很好,涌现了像天瑞仪器这样生机勃勃的国产厂商。期望通过合作,促进国产仪器的发展、提高民族产品的知名度。并希望有越来越多的人采购国产仪器。 当天,黄业茹主任一行在刘召贵博士的陪同下依次参观天瑞环保仪器展厅、化学实验室、前处理实验室、液体及固体样品库、生产车间、品质监管线、研发实验室等区域。他们在参观中表示:“作为仪器厂商,能有自己的化学及前处理实验室,这让我们很意外。同时,规范、强大的生产管理、品质监管及研发队伍,也使人印象深刻。” 7月23至26日,《空气和废气中颗粒物测定-XRF法标准》开题验证试验工作展开。验证试验由吴升海博士和姚栋梁博士主持,国家环境分析测试技术研究室主任李玉武博士全程参与指导。 目前,实验数据正在分析中,结果将呈至国家环境分析测试中心,并将据此,对《空气和废气中颗粒物测定-XRF法标准》进一步完善。http://ng1.17img.cn/bbsfiles/images/2011/07/201107281540_307276_2090336_3.jpg 洽谈会现场http://ng1.17img.cn/bbsfiles/images/2011/07/201107281541_307277_2090336_3.jpg 刘召贵博士在洽谈会议上作报告http://ng1.17img.cn/bbsfiles/images/2011/07/201107281542_307278_2090336_3.jpg 李玉武博士(右一)与吴升海博士(左一)共同参与标准验证试验
本文建立高效液相色谱法测定莱阳梨止咳颗粒中盐酸麻黄碱的含量。采用高效液相色谱法。色谱柱:岛津VP-ODS C18 (150mm×4.6mm, 5μm);流动相:乙腈-0.1%磷酸溶液(5∶95);检测波长:205nm;流速:1mL/min;柱温:室温。样品浓度在6.752~40.512μg/mL 范围内,浓度与峰面积比有良好的线性关系,重现性RSD=1.1%,稳定性RSD=1.3%,平均回收率为97.6%。该方法稳定、可靠,可作为该制剂的质量控制。
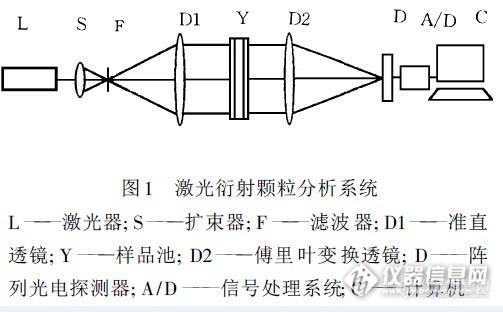
当代激光颗粒分析技术的进展与应用任 中 京( 济南微纳颗粒仪器股份有限公司 济南 250022)摘 要:简要介绍了当代激光颗粒分析技术的最新主要的进展。内容涉及测试原理的发展、仪器结构的改进、数据处理技术的突破、多次散射的处理、样品分散系统的多样化、颗粒形状对测试的影响、颗粒散射模型、工业在线应用等一系列理论和应用问题。关键词:激光,粉体,颗粒,散射,测试1 前言著名物理学家费曼曾说: 假如由于某种大灾难,所有的科学知识都丢失了,只有一句话传给下一代,那么怎样才能用最少的词汇来表达最多的信息呢? 我相信这句话是原子的假设,所有的物体都是用原子构成的 。”可见物质组成在人类文明中具有多么重要的意义。20 世纪,人们对于宏观与微观的物理世界已经有了相当深入的了解,但是对于微观粒子到宏观物体之间的大量物理现象却知之甚少。颗粒正是二者之间的中介物。如大颗粒主要表现为固体特性。随着颗粒变小,流动性明显增强,很像液体;颗粒进一步变小,它将像气体一样到处飞扬了;颗粒尺度再小,它的表面积则迅速增大,表面的分子所处状态与大颗粒完全不同,颗粒的性质将发生突变,显示出某些令人震惊的量子特性! 现在, 世界上许多优秀的科学家正在这个介观领域辛勤耕耘,大量具有特殊性能的材料将在这一领域诞生。导致颗粒性质发生如此变化的第一特征是它的大小。颗粒大小在人们的生活和生产中也非常重要。如水泥颗粒磨细些,水泥早期强度将明显提高;药品粒度越细,人体对它的吸收越好;磁性记录材料越细,存储密度越高。这样的例子不胜枚举。因此,颗粒超细化已经成为提高材料性能的重要手段。颗粒大小测定受到人们重视也就不足为奇了。人们为了测定颗粒大小,几乎采用了可以想到的一切办法。由于篇幅所限,本文只介绍激光颗粒分析技术的概况。2 激光怎样测量颗粒大小激光测量颗粒大小的方法有多种,其中包括光散射、光衍射、多普勒效应、光子相关谱、光透法、消光法、光计数器、全息照相等,本文所说的激光颗粒分析专指通过检测颗粒群的散射谱分布,分析其大小及分布的激光散射( 衍射) 颗粒分析技术。众所周知,一束平行激光照射在颗粒上,将发生著名的夫琅禾费衍射,使用傅里叶变换透镜汇集衍射光,在透镜后焦面可得到此颗粒的衍射谱。如果颗粒是球体,则衍射谱是著名的Airy 图形,中心的Airy 斑直径与颗粒直径成反比。若将一同心环阵光电探测器置于后焦面用于衍射谱的检测,再配以信号处理系统, 即构成基本的激光衍射颗粒分析系统 (见图1) 。http://ng1.17img.cn/bbsfiles/images/2015/12/201512221524_579009_3049057_3.jpg当光束中无颗粒存在时,光会聚在探测器中心; 当小颗粒进入光束时, 探测器的光强分布较宽;当大颗粒进入光束时,探测器光强分布较窄。如果进入光束检测区的是具有一定粒度分布的颗粒群, 则探测器的输出为全部颗粒衍射谱的线性叠加,使用反演技术可根据衍射谱反求被测颗粒群的粒度分布 。激光衍射颗粒分析系统适用于粒度大于激光波长很多的颗粒,测量范围大约在6Lm 以上,测量上限决定于透镜焦距,已知最大可测到2000Lm.激光颗粒分析系统的优点是非常突出的,其中包括(1) 测量速度快,其他方法无法比拟;(2)测量过程自动化程度高,不受人为因素干扰,准确可靠;(3)衍射谱仅与颗粒大小有关,与颗粒的物理化学性质无关,因此适用面极广。3 从衍射到散射使用衍射原理的激光颗粒分析系统的主要缺点是在小颗粒范围测量误差很大,特别是无法测量亚微米颗粒的大小。随着颗粒技术的进步,颗粒粒度迅速向超细发展,夫琅禾费衍射已不能满足测试要求,必需采用更精确的Mie 理论。http://ng1.17img.cn/bbsfiles/images/2015/12/201512221525_579010_3049057_3.jpgMie 散射理论是球形颗粒对单色光的散射场分布的严格解析解。夫琅禾费衍射是Mie 散射理论在特定条件下的近似。Mie 散射理论指出,当颗粒直径比入射光波长小得多时,颗粒的前向散射与后向散射场分布对称;当颗粒直径与入射光波长近似时,前向散射比后向散射强,且散射场关于入射光轴呈周期分布;当颗粒直径比入射光波长大得多时,颗粒将只有前向散射场,这正与夫琅禾费衍射理论一致(见图2) 。由此可见,Mie 散射理论比夫琅禾费衍射理论适用范围更广,更精确。为了适应小颗粒散射谱的测量,光路也发生了重大变化,原平行光路由会聚光路取代。颗粒样品由置于透镜前改为透镜之后,可接收的散射角达到70b。经改进的颗粒分析新光路测量范围从0.1um 至数百um,只要改变样品位置即可方便地调节测量范围,不必更换透镜 。至此,Mie 散射理论正式担当了颗粒分析的主角。4 多重散射激光散射颗粒分析在原理上要求被测颗粒无重叠随机分散在与光路垂直的同一平面内。但是这一要求在实际上很难做到,例如干粉从喷嘴喷出往往呈三维分布,前面的颗粒使平行激光发生散射,散射光遇到后面的颗粒再次散射,此过程经历多次,散射谱分布大大展宽,这种现象称为多重散射。可以证明,N 次散射光场的复振幅是单次散射光场的复振幅的N重卷积。颗粒分布得越厚,散射谱展宽越严重,颗粒分析结果将严重地向小颗粒偏移。为了抑制多重散射,人们曾采用了多种办法。我国学者分析了多重散射与颗粒浓度的关系,发现颗粒三维分布时仍存在最佳衍射浓度,在此浓度下,多重散射可以得到有效抑制。颗粒分布越厚,最佳衍射浓度则越小。在此理论指导下,我国研制的干粉激光颗粒分析仪,其测量结果可以同湿法激光颗粒分析仪相比。5 反演——追求真实的努力我们的测量对象很少有单一粒径的颗粒集合,往往是有一定粒度分布的颗粒群。我们所测得的谱分布是由颗粒分布函数为权重的颗粒散射谱分布对所有粒径的积分。在颗粒分析中的反演运算即通过所测谱分布反求粒度分布(颗粒的散射谱分布作为理论已知)。反演正确与否直接关系到此技术的成败。本文不想全面论述反演技术,只简要介绍两种反演思路。流行的一种方法是先假定被测颗粒粒度服从某种分布函数( 如正态分布、对数正态分布、R - R 分布等,然后叠代求取分布参数。如果预先的假定是错的,那么反演结果必错。怎样才能获得真实可靠的结果呢? 我国研究人员发展了一种无约束自由拟合反演技术,即对粒度分布函数不作任何约束,令每一权重因子独立地逼近最佳值。此技术已在仪器上应用并取得良好效果,提高了颗粒大小分辨率,保证了反演结果的真实可靠性。此技术在其他场合也有应用价值。6 大小与形状有关吗?通常认为物体的大小与物体的形状是互不相关的两个概念。近期关于颗粒学的研究表明,颗粒大小的表征不仅与颗粒形状有关,而且与颗粒测试的方法有关,这恐怕是人们预料不到的。以沉降法为例来说明。在重力场中,某非球形颗粒A 的最终沉降速度与另一同质球体B的最终沉降速度相同,则定义颗粒A 的粒径即为颗粒B 的球体直径,称为沉降粒径。二者实际体积并不相同。与此相反,体积相同的两颗粒,若形状不同,一为球体另一为非球体,则其沉降粒径也不同。由此看来颗粒大小与形状有关。与沉降法类似,激光散射法所测粒径也与形状有关。截面积相同的两颗粒,非球体的衍射谱比球体的谱宽。若用球体衍射谱度量非球体,则测试结果偏小。为了解决这种矛盾,我国学者引入椭圆颗粒衍射模型,即取非球体颗粒的最小外圆直径为长轴,取其最大内圆直径为短轴,所作椭圆即为该颗粒的椭圆模型。颗粒的球体模型发展到椭圆模型是颗粒学的一个进步,椭圆模型引入的实质就是承认颗粒大小与颗粒形状有关,并把形状因素引入大小度量的范畴。椭圆模型的引入,为激光颗粒分析用于非球形颗粒奠定了理论基础,并有效地提高了测量精度。7 从实验室到工业生产第一线事实上颗粒测试生产线早已需要一种颗粒在线检测设备。例如粉磨设备的主要功能是将原料磨细,因此颗粒大小就成为粉磨工艺的首要检测指标,但是无论是沉降法还是库尔特法,无论是图像法还是超声波法,均难担此重任。目前人们只能靠检测磨机负荷与监听磨机发出的声音来判断它的工作状态,至于产品粒度则需数小时一次间隔取样,到试验室分析,再返回现场调整磨机,由于检测不及时,导致产品过粗或过粉磨现象司空见惯,造成的浪费无法计算。现在,激光颗粒分析技术的出现与成熟,为颗粒在线测试提供了可能。激光颗粒分析技术除前面谈到的许多优点外,还有一些优点尚未引起人们的注意:(1)它可用于运动颗粒群的实时颗粒分析;(2)它不但适用于液体中的颗粒,也适用于气体中的颗粒。所有这些优点都注定了这种测试方法必定要在现代化的颗粒生产线担任在线粒度测试的主角。此技术在粉磨系统的应用必将改变磨机的控制模式,磨机将发挥出更大的潜力,能耗也将得到最大限度的节约。我国在气流粉碎机方面的粒度在线测控研究工作业已取得可喜的成果。预计不久,选粉、造粒、喷雾、干燥、结晶等许多工艺过程都将由激光颗粒分析仪担当在线分析的重任。到那时,此种技术的潜力才可得到较为充分的发挥。8 结束语激光颗粒分析技术的研究从70 年代起步,到今天才不过20 年的时间,它已经在测量精度、测量速度、分辨能力、动态检测能力等方面远远超过传统分析方法,在世界许多实验室与生产企业应用表现出无可比拟的优越性,越来越多的产品正在选择激光颗粒分析技术作为产品检验标准。此种
长达10年的上海大气颗粒物源解析已取得初步成果,“常态源解析初步揭示PM2.5来源,得出空气中PM2.5本地人为污染排放贡献占八成,交通和工业是重头。其中 初步研究表明,上海市PM2.5的主要来源为:机动车船等移动源占25%;石化、化工、工业喷涂、钢铁和建材等工业工艺过程占15%;工业锅炉、工业炉窑占11%;电站锅炉占10%;建筑工地、道路和堆场扬尘等占10%;干洗、餐饮和民用涂料等生活面源占5%;秸秆燃烧、化肥使用和畜禽养殖等农业源占4%;区域影响占20%。 在以上PM2.5的的主要来源中,机动车尾气污染不容忽视,大量的柴油车在道路行驶过程中会产生PM2.5,司机不良驾驶习惯如急加速、急减速还会排放更多的PM2.5。同时上海市还有20多万辆的高污染黄标车在道路上行驶,这些车排放的大气污染物是国四排放标准车辆的二十到三十倍,加速淘汰高污染车辆势在必行。 基于此,上海制定了180多项有针对性治理措施。上海市环境监测中“为了定性或定量识别大气颗粒物来源,上海逐步构建大气污染源排放清单与源谱,探索颗粒物来源解析技术方法,在常态源解析和重污染快速源识别两个方面均取得进展,并在治污中得到有效应用。
现在用在线颗粒度分析仪检测颗粒度变化的单位越来越多,都有哪些厂家可以做这样的仪器?
有谁做过煤炭颗粒亚甲蓝吸附值的测定?希望给点经验。
颗粒的测量颗粒测定需要采用稀释系统。对于颗粒的测量,美国环境保护总局(EPA)规定用全流式稀释风道,欧洲则允许使用分流式系统。全流式稀释风道占用面积大、设备投资大,只适用于固定实验室。在实际测量过程中,美国铁路也有使用分流式系统进行测试的(如:GM-EMD)。
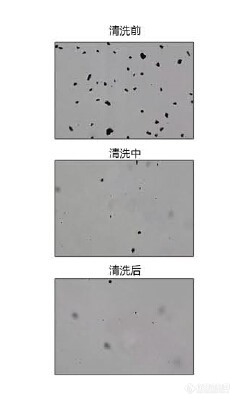
自清洗样品窗在动态颗粒图像技术的应用一、 从静态图像仪到动态图像仪早期的颗粒图像仪都是静态颗粒图像仪,基本上是基于显微镜设备改装的观测设备,制作静态样品,虽然在一定程度上解决了颗粒样品的形貌分析统计问题,但是也表现出了其固有的弱点,即因其参与观测统计的颗粒数量少,导致数据的代表性差。人为误差较大。因此在上世纪90年代末国外就开始进行动态颗粒图像仪的研制,英、法德等国家均推出过动态颗粒图像测试设备。而在本世纪初,国内的上海理工、天津海洋研究所等机构也开始探索颗粒动态测试的有效方法。直到2007济南某厂家首次正式面向市场推出真正意义上国内第一台动态颗粒图像分析仪Winner100。中国才真正具有了动态颗粒图像分析能力。二、 动态图像技术分析对微小颗粒而言,成像光路系统放大倍率越大,其景深也就越小,这一点严重制约动态颗粒图像仪的发展,如何将流动中的颗粒约束到一个平面上,这是动态颗粒图像仪最关键部分。目前国外现有的比较成熟的方式借鉴了细胞测量中的流体聚焦技术----鞘流技术,即将待测颗粒样品流入鞘液中,鞘液对其进行约束,形成一个一个从而获得清晰的颗粒图像。这种技术能够解决颗粒聚焦问题,但是其制备鞘液比较复杂,成本也很高,测量时间也较长,而且鞘液中的颗粒数量仍然不能够太多,因此对于颗粒测试的代表性仍然不强。关键部件鞘流池如果有大的颗粒进入很容易发生堵塞现象,清理疏通也都很费时费力。以国外很多粒度仪厂家也多采取这种实用价值有限的测试技术。近年国内厂家推出一种新型技术,即以流体力学的原理,使用液流的压力将颗粒约束在样品窗表面,使其基本在一个焦平面上运动,使成像效果显著提高。但是问题随之而来,在样品窗表面运动时,经常有颗粒粘连在表面上,越积越多无法处理。因此,此方法的使用价值也大打折扣。2014年济南微纳颗粒推出了一款带超声波自清洗装置的样品窗,才真正解决了这种颗粒在样品窗上粘连的问题,使其实用化程度大大提高,现在在碳化硅、氧化铝等磨料相关等行业已经广泛开始使用,并得到了用户的高度认可。三、 自清洗样品窗技术在以往的动态图像仪中,样品窗污染就会造成测试结果的准确性差。因此样品窗必须每隔一至两周就必须拆卸下来清洗,去除附着在上面的颗粒残留,非常麻烦,而且有的样品自身带有粘性或者静电的,甚至在测试过程中就会粘连到样品窗上,严重影响测试结果。济南微纳推出的可以进行自清洗的样品窗,彻底解决了以上问题,大大减少了样品窗的清洗频次,增加了样品窗寿命,有的甚至可以终生不必拆洗。 自清洗样品窗技术已经应用在微纳的Winner100D动态图像仪、Winner219动静态双模式全自动图像仪上,并解决了样品窗清洗问题。并且自清洗样品窗技术还可以应用在湿法激光粒度仪上,微纳也将进一步自清洗样品窗技术广泛的推广应用,为推动中国颗粒测试事业的发展尽最大努力。 http://ng1.17img.cn/bbsfiles/images/2015/11/201511201552_574512_3049057_3.png
颗粒测试技术的进展与展望摘 要:本文简述了当今颗粒测试技术六个方面的进展,对颗粒测试技术的近期发展趋势作了简短的展望,提出了七个颗粒测试领域需要统一认识的基本问题,对促进颗粒测试技术发展提出了几点建议.关键词:颗粒测试;技术进展;发展趋势;基本问题;知识产权1 前 言随着颗粒技术的发展,颗粒测试技术已经受到广泛的关注与重视. 本文就目前颗粒测试领域的新进展,谈一点个人的浅见,请各位指教. 本文谈及的问题有:颗粒测试技术进展、颗粒测试技术展望、颗粒测试的基本问题和促进颗粒测试技术发展的几点建议.2 颗粒测试技术进展近年来颗粒测试技术进展很快,表现在以下几个方面:1) 激光粒度测试技术更加成熟,激光衍射/散射技术,现在已经成为颗粒测试的主流. 其主要特点:测试速度快,重复性好,分辨率高,测试范围广得到了进一步的发挥.激光粒度分析技术最近几年的主要进展在于提高分辨率和扩大测量范围. 探测器尺寸增加,附加探头的使用扩大了测量范围;多种激光光源的使用、多镜头、会聚光路、多量程、可移动样品窗的使用提高了分辨率,采样速度的提高则进一步改善了仪器的重复性. 英国马尔文公司GM2000系列激光粒度仪采用高能量蓝光辅助光源和汇聚光学系统,测量范围达到0.02?2000微米,不需更换透镜. 贝克曼库尔特公司采用多波长偏振光双镜头技术将测量范围扩展到0.04?2000微米.代表了当前的先进水平. 国产的激光粒度仪在制作工艺和自动化程度上尚有欠缺,但大多数在重复性准确度方面也达到了13320国际标准的要求. 目前激光粒度分析仪在技术上,已经达到了相当成熟的阶段.米氏理论模型可以提高仪器的分辨率,但是需要事先了解被测样品的折射率和吸收系数,才可能获得正确的结果.测试结果的优劣不仅取决于测试系统和计算模型,更加取决于样品的分散状态.激光粒度仪对样品的分散要求是,分散而不分离. 仪器厂家应更加注意样品分散系统设计. 尽量避免小颗粒团聚,大颗粒沉降,大小颗粒离析,样品输运过程的损耗,外界杂质的侵入. 对于不同样品选用不同的分散剂和不同的分散操作应该引起测试者的注意.任何原理的仪器测试范围都不是可以无限扩展的. 静态光散射原理的激光粒度分析向纳米颗粒的扩展和向毫米方向的扩展极限值得探讨. 毫米级的颗粒只需光学成像技术就可以轻易解决的测量问题采用激光散射原理则并不是优势所在.2) 图像颗粒分析技术东山再起图像颗粒分析技术是一种传统的颗粒测试技术,由于样品制备操作较繁琐、代表性差、曾经作为一种辅助手段而存在,他的直观的特点没有发挥出来.为了解决采样代表性问题,有人使用图像拼接技术或者多幅图像数据累加技术可以有效提高分析粒子数量,采用标准分析处理模式的图像仪则可以将操作误差减小,这些改进取得了一定的效果.最近几年动态图像处理技术的出现使传统度颗粒图像分析仪备受关注,大有东山再起之势. 动态图像处理的核心是采用颗粒同步频闪捕捉技术,拍摄运动颗粒图像,因此减少了载玻片上样品制备的繁琐操作,提高了采样的代表性,而且可用于运动颗粒在线测量. 这就大大扩展了图像分析技术的应用范围和可操作性. 荷兰安米德公司的粒度粒形分析仪是有代表性的产品。它采用CCD+频闪技术测颗粒形状、采用光束扫描技术测颗粒大小。可测最大粒径为6毫米。如果颗粒在光学采样过程不发生离析现象,此种仪器在微米与毫米级颗粒测量中可能会得到广泛的应用.颗粒图像分析技术需要解决的另一个问题是三维测量. 动态颗粒图像采集由于颗粒采集的各向同性因此可以解决在载波片上颗粒方位的偏析问题,但是仍然无法解决如片状颗粒厚度问题. 厚度测量对于金属颜料,云母、特种石墨都是一个急需解决的实际问题.3) 颗粒计数器不可替代颗粒本身是离散的个体,因此对颗粒分级计数是一种最好的测量方法. 库尔特电阻法在生物等领域得到广范应用已经成为磨料和某些行业的测试标准. 但是他受到导电介质的限制和小孔的约束,在某些行业推广受到阻力.最近光学计数器在市场上异军突起,他将在高精度和极低浓度颗粒测量场合发挥不可替代的作用. 美国Haic Royco 公司颗粒计数器/尘埃粒子计数器是才进中国不久的老产品;美国PSS(Particle Sizing Systems)公司采用单粒子光学传感(SPOS)技术生产的系列仪器可用于湿法、干法、油品等各种场合的颗粒计数。国内颗粒计数器的研究工作起步并不晚,但是除了欧美克的电阻法计数器外,尚未见光学计数器商业化的产品。4) 纳米颗粒测试技术有待突破纳米颗粒测试越来越受到重视.电镜是一种测试纳米颗粒粒度与形态最常用的方法.电镜样品制备对于测试结果有重要影响,北京科技大学在拍摄高质量电镜照片方面作了出色的工作. 由于电镜昂贵的价格和严格的使用条件,以及取样代表性问题,电镜在企业推广不是最佳选择.根据动态光散射原理设计的纳米级颗粒测试技术是一种新技术,近年来获得了快速发展.马尔文,布鲁克海文、贝克曼库尔特等公司提供了优秀的商品,马尔文公司已将动态光散射的测量范围扩展到亚纳米范围,HPPS高性能高浓度纳米粒度和Zeta电位分析仪测试范围0.6-6000纳米,可以测量大分子真溶液粒径。国内开展此项技术研究的单位日益增多,上海理工大学、浙江大学、北京大学、清华大学、济南大学等许多高校都有学者和研究生在做工作. 数字相关器仍然是制约国产动态光散射仪器的瓶颈技术,如果数字相关器问题得到解决,中国自己的动态光散射纳米粒度仪出现在市场上将不会太远.X射线的波长比纳米还要短,因此X射线小角散射是一种测量纳米颗粒的理想方法,(类似于激光衍射原理)国外有商品仪器. 国内,此方法已经列入国家开发计划,国家钢铁研究总院对此方法研究已经作了大量工作,但是尚未见商品问世.5) 光子相关技术独树一帜动态光散射原理纳米颗粒测试采用的技术主要是光子相关谱,光子相关技术是一种70年代兴起的超灵敏探测技术,他根据光子信号的时间序列的相关性检测被测信号的多普勒频移或时间周期性,比通常的光谱仪分辨率高一个数量级,因此此技术也被用于颗粒运动速度的测定和其他场合. 上海理工大学浙江大学利用此原理已经研制成功在线用的颗粒粒度与颗粒流速的探针. 它可用于物料管道内部检测物料的平均大小和物料的流速. 对于在线控制具有指导意义。有报道称使用光子探测技术可以对高压空气喷嘴中的颗粒计数,说明颗粒测试正在向更加精密更加灵敏的方向发展.6) 颗粒在线测试技术正在兴起







 我要推广仪器
我要推广仪器
 下载APP
下载APP