土壤活化剂是一个通用术语,用来描述可以添加到土壤中以改善土壤结构和提高植物生产力的各种产品。土壤是粘土或沙子、有机物质、有益细菌和真菌以及氧、氢和碳等化学物质的复杂混合物。随着植物的生长它们会吸收化学物质,土壤活化剂的作用是通过替换这些成分来恢复土壤的质量。土壤活化剂可以解决土壤板结,它可以分解土壤中有机物质,并增加土壤内有效养分,促进植物营养吸收,使植物生长的更加健康,并在一定程度上的保护植物根。

原实验用SYS305无铅焊膏用助焊剂许多性能不够完善。焊后焊点不够饱满、光亮,周边有锯齿状回缩等表面缺陷。并且在实际板级封装的印刷过程中焊膏的粘度太高,容易粘板,不能进行连续印刷,并且不能在封装版上润湿。针对这些缺点本实验对SYS305无铅焊膏用活化剂配方进行了调整改善。活化剂是助焊剂的关键组分。而在活化剂组分中,除了松香外,还有有机卤化物、有机酸和胺类可作为活化剂。目前,广泛使用烃基酸、芳香酸和羧酸等有机酸作活化剂,例如丁二酸、已二酸、戊二酸、苹果酸、谷氨酸、柠檬酸、酒石酸、苯甲酸和山梨酸等。本次实验活化剂选用A酸、B酸、C酸,通过调整它们子之间的复配比例和在助焊剂中的含量来进行实验。然后将复配后的活化剂溶于一定质量的溶剂中制成助焊剂,冷却后制备焊膏做铺展实验,以铺展的情况为评定指标来判定助焊剂性能的好坏。1、调整几种活化剂之间的复配比例A酸、B酸、C酸复配实验,命名为C组,调配6种助焊剂进行测试。配方如表1所示。表1 C组活化剂配方配方号溶剂总量(g)A酸(g)B酸(g)C酸(g)溶液总量(g)C140.710.170.1210C240.50.040.4610C340.360.480.1610C440.240.190.5710C540.130.790.0710C640.040.40.5610对C组进行铺展测试,铺展测试情况见图1。测试结果见表2。http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif图1 C组铺展测试情况http://ng1.17img.cn/bbsfiles/images/2017/10/2015092809203996_01_3042675_3.png表2 C组铺展测试结果配方号铺展情况铺展面积(mm2)C1边缘有回缩、飞溅,焊点有腐蚀、气孔85.306C2铺展面积较大,但边缘有飞溅90.054C3焊点饱满84.564C4腐蚀比较严重,存在残留物82.438C5回缩严重81.743C6回缩严重,存在残留物83.771通过铺展图对比,C组铺展面积都比较大,C3焊点饱满光亮,没有回缩、飞溅,铺展面积相对较大。2、调整活化剂占助焊剂总量的百分比根据活化剂对助焊剂的影响,调整活化剂在助焊剂中的百分比。在所用活化剂配比为C3组比例的基础上,调整占助焊剂总量的百分比,命名为D组,调配7种助焊剂进行测试。以D0组为对比。配方如表3所示。表3 D组活化剂配方配方号百分比(%)A酸(g)B酸(g)C酸(g)溶液总量(g)D0000010D151.65[align=cent
网上很多人说土壤活化剂是智商税,是真的吗?大家有用过此类产品吗?
C18固相萃取小柱的活化剂、洗脱试剂(如二氯甲烷、乙酸乙酯、甲醇等)必须用色谱醇的吗?用优级纯或分析纯试剂是不是可以?
你好!我想请问一下,我有镉电极和铅电极,头次使用但现因没有活化剂对它们进行活化,我将要用它们做电解阴极,如我不活化是否有影响?
Q1.引物是如何合成的?目前引物合成基本采用固相亚磷酰胺三酯法。DNA合成仪有很多种,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。(1) 去保护:加入Deblocking脱去碱基上5'- OH的保护基团DMT,获得游离的5'- OH;(2) 耦合:同时加入活化剂和新的碱基,新的碱基5'-OH仍然被DMT保护,3'端被活化与溶液中游离的5'-OH发生耦合反应;(3) 封闭:耦合反应中极少数5'- OH没有参加反应,用封闭试剂终止其后继续发生反应;(4) 氧化:加入氧化剂使其由核苷亚磷酸酯形成更稳定的核苷磷酸酯。Q2.引物合成后如何处理?切割与脱保护基:将合成好的寡核苷酸链从支持物上化学切割下来。常用新鲜的浓氨水来裂解CPG与初始核苷之间的酯键。断裂下来的寡核苷酸带有自由的3'羟基。纯化:根据所合成寡核苷酸的组成和应用来选定纯化的方法。常用的纯化方法有:C18、OPC、PAGE和HPLC。定量:根据寡核苷酸在260nm处的紫外吸收来定量。储存:分装抽干。Q3.需要什么级别的引物?根据实验需要,确定订购引物的纯度级别。应用引物长度要求纯度级别要求一般PCR扩增60 basePAGE诊断PCR扩增 40baseHEPD, PAGEDNA测序20base左右HEPD亚克隆,点突变等根据实验要求定HEPD, PAGE,HPLC基因构建(全基因合成)根据实验要求定HEPDPAGE反义核酸根据实验要求定HEPDPAGE修饰引物根据实验要求定PAGE, HPLC Q4.引物的质量是不是跟序列有关?四种碱基的性质和各自保护基的性质都有差别。所以合成难度是不一样的。难度最大的当属GC重复多的和序列中还有多个连续的G的引物。尤其对于后者,国内公司一般都做不了20个G以上的引物。实验证明,如果引物中有超过三个连续G的结构,传统方法得到的产物质量就会开始下降。而且目前通用的脱盐、OPC和PAGE方法都无效。鼎国昌盛生物公司拥有的HEPD专利技术能够克服高GC或者高G含量引物的合成和纯化障碍,对普通引物、高GC引物和无论长短的oligo d(G)都能得到同样的非常好的结果。Q5.需要合成多少OD数?根据实验目的确定。一般PCR扩增,2 OD引物,可以做500-1000次50ul标准PCR反应。如果是做基因拼接或退火后做连接,1 OD就足够了。Q6.如何计算引物的浓度?引物保存在高浓度的状况下比较稳定。一般情况下,我们建议将引物的浓度配制成100pmol/ul,称为保存浓度,而引物的工作浓度一般配制成10-50pmol/ul。加水的体积(微升)可直接参照合成报告单上推荐的体积,也可按下列方式计算:V (微升)= OD数x 33 x 10000 /引物的分子量引物的分子量可以从合成报告单上获得。注意:1 OD260= 33 ug/ml。Q7.如何计算引物的Tm值?引物设计软件都可以给出Tm,与引物长度、碱基组成及所使用缓冲夜的离子强度有关。长度为25base以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10 + 0.41 (GC%) - 600/size**公式中,Size =引物长度。
什么是活性炭 活性炭是一类具有发达的孔隙结构、很大的比表面积和极强的吸附性能的含碳物质。是以炭化料为原料进一步经过850~900℃的高温,同时加入大量的水蒸气作为活化剂处理得到的产物;每克活性炭的吸附面积相当于八个网球埸之多。其结构为炭形成六环物堆积而成,由于六环炭的不规则排列,造成了活性炭多微孔体积及高表面积的特性。 采用现代技术制备的活性炭,它的孔隙比孔容积可超过2 cm3/g,比表面积可达3000 m2/g以上,优异的吸附-解吸性能使它被称为“万能的吸附剂”。活性炭有哪些分类一、按原料分类常见的活性炭材质: 1、天然高分子材料 天然高分子材料主要分为两大类: A、植物类:果壳(杏壳、椰壳、核桃壳、山桃核、橄榄壳等)、木材(锯末、木屑等)、秸秆等 B、植物化石类:无烟煤、烟煤、褐煤 2、合成材料 合成材料主要是:石油焦、沥青、聚丙烯腈、废旧轮胎等 3、再生材料 因为活性炭在发生物理吸附时具有高度的可逆性,所以用过的活性炭经过再生后还能使用,以此为原料制造的活性炭为再生炭 目前市场常见的有果壳、木质、煤质三大类活性炭。二、按制造方法分类: 1、物理法 用水蒸气、二氧化碳或两者混合气体(活化剂为气体)为活化剂的气体活化方法 2、化学法 用酸、碱、盐(活化剂为固体)等化学品为活化剂的活化方法——主要活化剂:氯化锌、磷酸、氢氧化钾等(主要用于高性能活性炭——中孔的制造) 3、物理化学法 先后或同时使用物理法活化剂及化学法活化剂进行活化的方法三、按外观形状分类: 1、颗粒(成型)活性炭 A、不定型(破碎)颗粒活性炭 B、成型活性炭:柱状、球形、块状 2、粉状活性炭(日本又称“素灰”) 粉状活性炭是指外观尺寸小于0.18mm(80目)的粒子占多数的活性炭四、按孔径大小分类: 1、大孔活性炭 大孔活性炭是指活性炭的孔径大于50nm的达到一定比例的活性炭 2、中孔活性炭 中孔活性炭是指活性炭的孔径大约在2~50nm的达到一定比例的活性炭 3、微孔活性炭 微孔活性炭是指活性炭的孔径小于2nm的达到一定比例的活性炭五、按用途分类: 1、液相方面的应用 2、气相方面的应用 3、催化剂及催化剂载体等方面的应用 4、处理土壤污染和作物培育中的应用 活性炭有哪些用途1、空气净化 2、污水处理场排气吸附 3、饮料水处理 4、电厂水预处理 5、废水回收前处理 6、生物法污水处理[
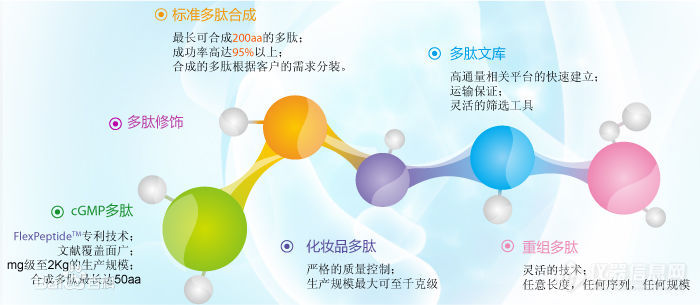
多肽合成技术主要采用多肽合成仪,以固相合成为反应原理,在密闭的防爆玻璃反应器中使氨基酸按照已知顺序(序列,一般从C端-羧基端 向 N端-氨基端)不断添加、反应、合成,操作最终得到多肽载体。固相合成法,大大的减轻了每步产品提纯的难度。为了防止副反应的发生,参加反应的氨基酸的侧链都是保护的。羧基端是游离的,并且在反应之前必须活化。固相合成方法有两种,即Fmoc和tBoc。由于Fmoc比tBoc存在很多优势,现在大多采用Fmoc法合成,但对于某些短肽,tBoc因其产率高的优势仍然被很多企业所采用。【请移步百度搜“[b]合肥国肽生物[/b]”即可】具体合成由下列几个循环组成:(1)去保护:Fmoc保护的柱子和单体必须用一种碱性溶剂(piperidine)去 除氨基的保护基团。(2)激活和交联:下一个氨基酸的羧基被一种活化剂所活化。活化的单体与游离的氨基反应交联,形成肽键。在此步骤使用大量的超浓度试剂驱使反应完成。循环:这两步反应反复循环直到合成完成。(3)洗脱和脱保护:多肽从柱上洗脱下来,其保护基团被一种脱保护剂(TFA) 洗脱和脱保护。多肽的分类多肽有生物活性多肽和人工合成多肽两种。1、生物活性肽生物活性肽(Bioactive Peptides ,BAP)是对生物机体的生命活动有益或是具有生理作用的肽类化合物,是一类相对分子质量小于6000Da , 具有多种生物学功能的多肽。生物活性肽具有多种人体代谢和生理调节功能,易消化吸收,有促进免疫、激素调节、抗菌、抗病毒、降血压、降血脂等作用,是当前国际食品界最热门的研究课题和极具发展前景的功能因子。2、人工合成多肽固相多肽合成方法(SPPS),由于其合成方便,迅速,成为多肽合成的首选方法,而且带来了多肽有机合成上的一次**,并成为了一支独立的学科——固相有机合成,固相合成的发明同时促进了肽合成的自动化。世界上第一台真正意义上的多肽合成仪出现在1980年代初期。基于将单个N-α保护氨基酸反复加到生长的氨基成份上,合成一步步地进行, 通常从合成链的C端氨基酸开始,接着的单个氨基酸的连接通过用DCC,混合炭酐, 或N-carboxy酐方法实现。Carbodiimide方法包括用DCC做连接剂连接N-和C-保护氨基酸。重要的是, 这种连接试剂促接N保护氨基酸自己炭基和C保护氨基酸自由氨基间的缩水,形成肽链, 同时产出N,N?/FONT-dyaylcohercylurea副产物。多肽合成方法1、酸酐法在多肽合成中,最初考虑应用酸酐要追溯到1881年Theodor Curtius对苯甲酰基氨基乙酸合成的早期研究。从氨基乙酸银与苯甲酰氯的反应中,除获得苯甲酰氨基乙酸外,还得到了BZ-Glyn-OH(n=2-6)。早期曾认为,当用苯甲酰氯处理时,N-苯甲酰基氨基酸或N-苯甲酰基肽与苯甲酸形成了活性中间体不对称酸酐。 大约在70年后,Theodor Wieland利用这些发现将混合酸酐法用于现代多肽合成。目前,除该方法外,对称酸酐以及由氨基酸的羧基和氨基甲酸在分子内形成的N-羧基内酸酐(NCA,Leuchs anhydrides)也用肽缩合。最后应该提到,不对称酸酐常常参与生化反应中的酰化反应。2、混合酸酐法有机羧酸和无机酸皆可用于混合酸酐的形成。然而,仅有几个得到了广泛的实际应用,多数情况下,采用氯甲酸烷基酯。过去频繁使用的氯甲酸乙酯,目前主要被氯甲酸异丁酯所替代。由羧基组分和氯甲酸酯起始形成的混合酸酐,其氨解反应的区域选择性依赖依赖于两个互相竞争的羰基的亲电性和(或)空间位阻。在由N保护的氨基酸羧酸盐(羧基组分)和氯甲酸烷基酯(活化组分,例如源于氯甲酸烷基酯)形成混合酸酐时,亲核试剂胺主要进攻氨基酸组分的羧基,形成预期的肽衍生物,并且释放出游离酸形式的活性成分。3、酰基叠氮物法酰基叠氮物法早在1902年就被引入到肽化学中,因此它是最古老的缩合方法之一。在碱性水溶液中,除了与酰基叠氨缩合的游离氨基酸和肽以外,氨基酸酯可用于有机溶剂中。与其他许多缩合方法不同的是,它不需要增加辅助碱或另一等当量的氨基组分来捕获腙酸。 长期以来,一直认为叠氮物法是唯一不发生消旋的缩合方法,随着可选择性裂解的氨基酸保护基引入,该方法经历了一次大规模的复兴。该方法的起始原料分别是晶体状的氨基酸酰肼或肽酰肼64,通过肼解相应的酯很容易得到。4、对称酸酐法Nα-酰基氨基酸的对称酸酐是用于肽键形成的高活性中间体。与混合酸酐法多肽合成相反,它与胺亲核试剂的反应没有模棱两可的区域选择性。但肽缩合产率最高,为50%(以羧基组分计)。虽然由对称酸酐氨解形成的游离Nα-酰基氨基酸可以和目标肽一起,通过饱和碳酸氢钠溶液萃取回收,但在最初,这种方法的实用价值极低。对称酸酐可以用Nα-保护氨基酸与光气,或方便的碳二亚胺反应制得。两当量的Nα-保护氨基酸与-当量的碳二亚胺反应有利于对称酸酐的形成,对称酸酐可以分离出来,也可不经纯化而直接用于后面的缩合反应。基于Nα-烷氧羰基氨基酸的对称酸酐对水解稳定,可采用类似上述纯化混合酸酐的方法进行纯化。[img=,690,300]https://ng1.17img.cn/bbsfiles/images/2019/04/201904221451156040_1751_3531468_3.jpg!w690x300.jpg[/img]我们主要提供:多肽合成、定制多肽、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。合肥国肽生物官网:http://www.bankpeptide.com欢迎咨询服务热线:17718122172;17718122684;17730030476;17718122397
C18固相萃取小柱,甲醇和水活化
多肽合成是一个重复添加氨基酸的过程,固相合成顺序一般从C端(羧基端)向 N端(氨基端)合成。固相合成法,大大的减轻了每步产品提纯的难度。为了防止副反应的发生,参加反应的氨基酸的侧链都是保护的。羧基端是游离的,并且在反应之前必须活化。固相合成方法有两种,即Fmoc和tBoc。由于Fmoc比tBoc存在很多优势,现在大多采用Fmoc法合成。【详情请咨询合肥国肽生物】(1)具体合成由下列几个循环组成:1. 去保护:Fmoc保护的柱子和单体必须用一种碱性溶剂(piperidine)去 除氨基的保护基团。2. 激活和交联:下一个氨基酸的羧基被一种活化剂所活化。活化的单体与游离的氨基反应交联,形成肽键。在此步骤使用大量的超浓度试剂驱使反应完成。循环:这两步反应反复循环直到合成完成。3. 洗脱和脱保护:多肽从柱上洗脱下来,其保护基团被一种脱保护剂(TFA) 洗脱和脱保护。(2)树脂的选择及氨基酸的固定将固相合成与其他技术分开来的最主要的特征是固相载体,能用于多肽合成的固相载体必须满足如下要求:必须包含反应位点(或反应基团),以使肽链连在这些位点上,并在以后除去;必须对合成过程中的物理和化学条件稳定;载体必须允许在不断增长的肽链和试剂之间快速的、不受阻碍的接触;另外,载体必须允许提供足够的连接点,以使每单位体积的载体给出有用产量的肽,并且必须尽量减少被载体束缚的肽链之间的相互作用。用于固相法合成多肽的高分子载体主要有三类:聚苯乙烯-苯二乙烯交联树脂、聚丙烯酰胺、聚乙烯-乙二醇类树脂及衍生物,这些树脂只有导入反应基团,才能直接连上(第一个)氨基酸。根据所导入反应基团的不同,又把这些树脂及树脂衍生物分为氯甲基树脂、羧基树脂、氨基树脂或酰肼型树脂。BOC合成法通常选择氯甲基树脂,如Merrifield树脂;FMOC合成法通常选择羧基树脂如王氏树脂。氨基酸的固定主要是通过保护氨基酸的羧基同树脂的反应基团之间形成的共价键来实现的,形成共价键的方法有多种:氯甲基树脂,通常先制得保护氨基酸的四甲铵盐或钠盐、钾盐、铯盐,然后在适当温度下,直接同树脂反应或在合适的有机溶剂如二氧六环、DMF或DMSO中反应;羧基树脂,则通常加入适当的缩合剂如DCC或羧基二咪唑,使被保护氨基酸与树脂形成共酯以完成氨基酸的固定;氨基树脂或酰肼型树脂,却是加入适当的缩合剂如DCC后,通过保护氨基酸与树脂之间形成的酰胺键来完成氨基酸的固定。(3)氨基、羧基、侧链的保护及脱除要成功合成具有特定的氨基酸顺序的多肽,需要对暂不参与形成酰胺键的氨基和羧基加以保护,同时对氨基酸侧链上的活性基因也要保护,反应完成后再将保护基因除去。同液相合成一样,固相合成中多采用烷氧羰基类型作为α氨基的保护基,因为这样不易发生消旋。最早是用苄氧羰基,由于它需要较强的酸解条件才能脱除,所以后来改为叔丁氧羰基(BOC)保护,用TFA(三氟乙酸)脱保护,但不适用含有色氨酸等对酸不稳定的肽类的合成。chang Meienlofer和Atherton等人采用Carpino报道的Fmoc(9-芴甲氧羰基)作为α氨基保护基,Fmoc基对酸很稳定,但能用哌啶-CH2CL2或哌啶-DMF脱去,近年来,Fmoc合成法得到了广泛的应用。羧基通常用形成酯基的方法进行保护。甲酯和乙酯是逐步合成中保护羧基的常用方法,可通过皂化除去或转变为肼以便用于片断组合;叔丁酯在酸性条件下除去;苄酯常用催化氢化除去。对于合成含有半胱氨酸、组氨酸、精氨酸等带侧链功能基的氨基酸的肽来说,为了避免由于侧链功能团所带来的副反应,一般也需要用适当的保护基将侧链基团暂时保护起来。保护基的选择既要保证侧链基团不参与形成酰胺的反应,又要保证在肽合成过程中不受破坏,同时又要保证在最后肽链裂解时能被除去。如用三苯甲基保护半胱氨酸的S-,用酸或银盐、汞盐除去;组氨酸的咪唑环用2,2,2-三氟-1-苄氧羰基和2,2,2-三氟-1-叔丁氧羰基乙基保护,可通过催化氢化或冷的三氟乙酸脱去。精氨酸用金刚烷氧羰基(Adoc)保护,用冷的三氟乙酸脱去。我们主要提供:多肽合成、定制多肽、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。合肥国肽生物官网:http://www.bankpeptide.com欢迎咨询服务热线:0551-62626599
引物篇1.引物是如何合成的?目前引物合成基本采用固相亚磷酰胺三酯法。DNA合成仪有很多种, 主要都是由ABI/PE 公司生产,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。亚磷酰胺三酯法合成DNA片段,具有高效、快速的偶联以及起始反应物比较稳定的特点。亚磷酰胺三酯法是将DNA固定在固相载体上完成DNA链的合成的,合成的方向是由待合成引物的3'端向5'端合成的,相邻的核苷酸通过3'→5'磷酸二酯键连接。第一步是将预先连接在固相载体CPG上的活性基团被保护的核苷酸与三氯乙酸反应,脱去其5'-羟基的保护基团DMT,获得游离的5'-羟基;第二步,合成DNA的原料,亚磷酰胺保护核苷酸单体,与活化剂四氮唑混合,得到核苷亚磷酸活化中间体,它的3'端被活化,5'-羟基仍然被DMT保护,与溶液中游离的5'-羟基发生缩合反应。 第三步,带帽(capping)反应,缩合反应中可能有极少数5'-羟基没有参加反应(少于2%),用乙酸酐和1-甲基咪唑终止其后继续发生反应,这种短片段可以在纯化时分离掉。第四步,在氧化剂碘的作用下,亚磷酰形式转变为更稳定的磷酸三酯。经过以上四个步骤,一个脱氧核苷酸被连接到固相载体的核苷酸上。再以三氯乙酸脱去它的5'-羟基上的保护基团DMT,重复以上步骤,直到所有要求合成的碱基被接上去。合成过程中可以观察TCA处理阶段的颜色判定合成效率。通过氨水高温处理,连接在CPG上的引物被切下来,通过OPC, PAGE等手段纯化引物,成品引物用C18浓缩,脱盐,沉淀。沉淀后的引物用水悬浮,测定OD260定量,根据定单要求分装。2.引物纯化方式有哪些,如何选择?◆ C18柱脱盐:有人称其为简易反相柱,它对DNA有特异性的吸附,可以被有机溶解洗脱,但不会被水洗脱,所以能有效地去除盐分。它不能有效去除比目的片段短的小片段。实际上,它是一种脱盐的作用。这种方法一般不会对普通PCR反应产生影响。对于需要用于测序、克隆的引物不能使用这个级别。 ◆ OPC纯化: OPC纯化是根据DNA保护基(DMTr基)和Cartridge柱中树脂间的亲合力作用的原理进行纯化目的DNA片段。OPC法纯化的DNA纯度大于95%。适用于40mer以下引物的纯化。◆ PAGE纯:PAGE纯化法是使用变性聚丙烯酰胺凝胶电泳,对DNA片段进行分离,然后从凝胶中回收目的DNA的方法。PAGE纯化法也是一种非常有效的DNA纯化方法,纯化后的DNA纯度大于95%,对长链Oligo DNA (大于50mer)的纯化特别有效。 ◆ HPLC纯化:HPLC纯化是使用高效液相色谱的原理,对DNA片段进行纯化。纯度可以大于99%。主要用于短链和修饰引物的纯化。该法的弱点是成本较高,批量生产效率不高。3.引物的OD数如何定量?答:引物合成引物OD数是这样测定的:用紫外分光光度计,波长260nm,石英比色杯,光程为1厘米,测定溶液的光密度。测定时溶液的光密度最好稀释到0.2-1.0之间。DNA干粉用一定体积的水充分振荡溶解以后,用1ml水稀释测OD值。需要根据稀释倍数换算出母液的OD值。4.需要什么级别的引物?答:引物常用的纯化方式C18脱盐,OPC纯化,PAGE纯化,HPLC纯化。根据实验需要,确定订购引物的纯度级别。应用 引物长度要求 纯度级别要求一般PCR扩增 45 base PAGE诊断PCR扩增 40base OPC, PAGEDNA测序 20base左右 OPC亚克隆,点突变等 根据实验要求定 OPC, PAGE,HPLC基因构建(全基因合成) 根据实验要求定 PAGE反义核酸 根据实验要求定 PAGE修饰引物 根据实验要求定 PAGE, HPLC5.最长可以合成多长的引物?答:引物越长,出现问题的概率就越大。我们合成过120base的引物,但是产率很低。除非需要,建议合成片段长度不要超过80mer,按照目前的引物合成效率,80mer的粗产品,全长(还不一定正确)引物的百分比不会超过40%,后续处理还有丢失很多,最后的产量是很低。6.需要合成多少OD数?答:根据实验目的确定。一般PCR扩增,2 OD引物,可以做200-500次50ul标准PCR反应。如果是做基因拼接或退火后做连接,1 OD就足够了。但是有些研究人员,就做几次PCR,但是却要5-10 OD。做全基因构建的引物都比较长,但是我们有些研究人员也要求高OD数。片段越长, 最后全长得率就越低,出错的几率就越大。超出需要之外的OD数要求,其实也是对社会资源的一种浪费,同时也从一个侧面反映了部分研究人员,特别是新手的自信心不足,总觉得需要重复多次才能成功。7.如何检测引物的纯度?答:实验室方便的作法是用PAGE方法。使用加有7M尿素的16%的聚丙烯酰胺凝胶进行电泳。取0.2-0.5OD的引物,用尿素饱和液溶解或引物溶液中加入尿素干粉直到饱和,上样前加热变性(95℃,2mins)。加入尿素的目的一是变性,二是增加样品比重,容易加样。600V电压进行电泳,一定时间后(约2-3小时),剥胶,用荧光TLC板在紫外灯下检测带型,在主带之下没有杂带,说明纯度是好的。如果条件许可,也可以用EB 染色或银染方式染色。8.如何计算引物的浓度?答:引物保存在高浓度的状况下比较稳定。引物一般配制成10-50pmol/ul。 溶解前您需要核对合成报告单和引物标签上的引物OD数是否一致。如果不一致,请和我们联系。我们可以根据生产记录查到实际产量是多少。一般情况下,我们建议将引物的浓度配制成50pmol/ul,加水的体积(微升)按下列方式计算:V (微升)= OD数*(乘)33 *(乘)*(乘)20000 / (除) 引物的分子量。引物的分子量可以从合成报告单上获得。如果需要配制成其他浓度,按上述公式换算。注意:1 OD260= 33 ug/ml.9.如何计算引物的Tm值?答:引物设计软件都可以给出Tm,引物长度,碱基组成,引物使用缓冲的离子强度有关。长度为25mer以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10 + 0.41 (%GC) – 600/size 公式中,Size = 引物长度。Tm的定义:Tm = Temperature at which 50% of a given oligonucleotide is hybridized to its complementary strand. In the absence of destabilizing agents, like formamide or urea, Tm will depend on 3 major parameters: The sequence: a GC-rich sequence has a higher melting temperature. The strand concentration: high oligonucleotide concentrations favor hybrid formation, which results in a higher melting temperature. The salt concentration: high ionic strength results in a higher Tm as cations stabilize the DNA duplexes. 10.引物(含修饰)的分子量是如何确定的?答:非修饰的引物的Molecular Weight在随引物提供的报告单上都有明确的标示。如果需要估计一个引物的分子量按每个碱基的平均分子量为324.5,引物的分子量=碱基数 x 碱基的平均分子量。或按下列公式计算MW= (NA * WA) + (NC * WC) + (NG * WG) + (NT * WT) +(Nmod * Wmod) +(Nx * Wx)+( Ni* Wi) +16* Ns– 62. NA, NG, NC, NT, Ni分别为引物中碱基A或G或C或T或I的数量,WA, WC, WG, W, Wi分别为引物中碱基A或G或C或T或I的分子量,Nmod,Wmod 分别为修饰基团的数目和分子量。对于混合碱基的分子量为混合碱基的分子量总合除以混合数,例如G+A混合的分子量为(313.21+329.21)/2 = 321.21。Ns为硫代数目,硫代每个位置增加分子量16。常规碱基分子量Base Molecular Weight A 313.21C 289.18G 329.21T 304.19I 314.2U 290.17常规修饰基团分子量5’-Biotin 405.45 3’-TAMARA 623.605’-(6 FAM) 537.46 3’-Dabsyl
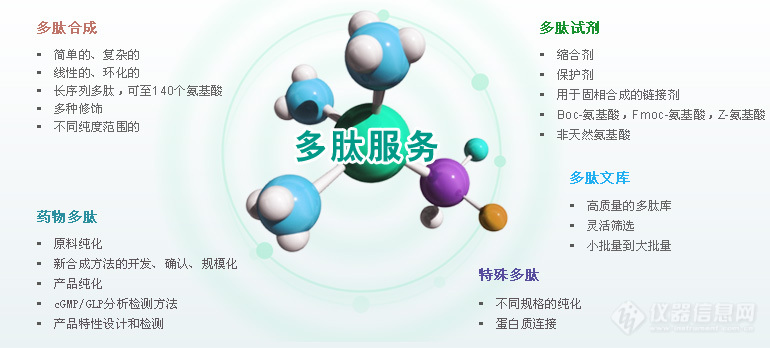
多肽合成又叫肽链合成,是一个固相合成顺序一般从C端(羧基端)向N端(氨基端)合成。过去的多肽合成是在溶液中进行的称为液相合成法。多肽的合成主要分为两条途径:化学合成多肽和生物合成多肽。请移步百度搜“合肥国肽生物”即可多肽合成的原理多肽合成就是如何把各种氨基酸单位按照天然物的氨基酸排列顺序和连接方式连接起来。由于氨基酸在中性条件下是以分子内的两性离子形式(H3+NCH(R)COO-)存在,因此,氨基酸之间直接缩合形成酰胺键的反应在一般条件下是难于进行的。氨基酸酯的反应活性较高。在100℃下加热或者室温下长时间放置都能聚合生成肽酯,但反应并没有定向性,两种氨基酸a1和a2的酯在聚合时将生成a1a2…、a1a1…、a2a1…等各种任意顺序的混合物。为了得到具有特定顺序的合成多肽,采用任意聚合的方法是行不通的,而只能采用逐步缩合的定向多肽合成方法。一般是如下式所示,即先将不需要反应的氨基或羧基用适当的基团暂时保护起来,然后再进行连接反应,以保证多肽合成的定向进行。式中的X和Q分别为氨基和羧基的保护基,它不仅可以防止乱接副反应的发生,还具有能消除氨基酸的两性离子形式,并使之易溶于有机溶剂的作用。Q在有的情况下也可以不是共价连接的基团,而是由有机强碱(如三乙胺)同氨基酸的羧基氢离子组成的有机阳离子。Y为一强的吸电子基团,它能使羧基活化,而有利于另一氨基酸的自由氨基,对其活化羧基的羧基碳原子进行亲核进攻生成酰胺键。由此所得的连接产物是N端和C端都带有保护基的保护肽,要脱去保护基后才能得到自由的肽。如果肽链不是到此为止,而是还需要从N端或C端延长肽链的话,则可以先选择性地脱去X或Q,然后再同新的N保护氨基酸(或肽)或C保护的氨基酸(或肽)进行第二次连接,并依次不断重复下去,直到所需要的肽链长度为止。对于长肽的多肽合成来说,一般有逐步增长和片段缩合两种伸长肽链的方式,前者是由起始的氨基酸(或肽)开始。每连接一次,接长一个氨基酸,后者则是用N保护肽同C保护肽缩合来得到两者长度相加的新的长肽链。对于多肽合成中含有谷氨酸、天冬氨酸、赖氨酸、精氨酸、组氨酸、半胱氨酸等等带侧链功能团的氨基酸的肽来说,为了避免由于侧链功能团所带来的副反应,一般也需要用适当的保护基将侧链基团暂时保护起来。多肽合成方法分类多肽的合成主要分为两条途径:化学合成多肽和生物合成多肽。化学合成主要是以氨基酸与氨基酸之间缩合的形式来进行。在合成含有特定顺序的多肽时,由于多肽合成原料中含有官能度大于2的氨基酸单体,多肽合成时应将不需要反应的基团暂时保护起来,方可进行成肽反应,这样保证了多肽合成目标产物的定向性。多肽的化学合成又分为液相合成和固相合成。多肽液相合成主要分为逐步合成和片段组合两种策略。逐步合成简洁迅速,可用于各种生物活性多肽片段的合成。片段组合法主要包括天然化学连接和施陶丁格连接。近年,多肽液相片段合成法发展迅速,在多肽和蛋白质合成领域已取得了重大突破。在多肽片段合成法中,根据多肽片段的化学特定性或化学选择性,多肽片段能够自发进行连接,得到目标多肽。因为多肽片段含有的氨基酸残基相对较少,所以纯度较高,且易于纯化。多肽的生物合成方法主要包括发酵法、酶解法,随着生物工程技术的发展,以DNA重组技术为主导的基因工程法也被应用于多肽的合成。多肽的固相合成多肽的合成是氨基酸重复添加的过程,通常从C端向N端(氨基端)进行合成。多肽固相合成的原理是将目的肽的第一个氨基酸C端通过共价键与固相载体连接,再以该氨基酸N端为合成起点,经过脱去氨基保护基和过量的已活化的第二个氨基酸进行反应,接长肽链,重复操作,达到理想的合成肽链长度,最后将肽链从树脂上裂解下来,分离纯化,获得目标多肽。1、Boc多肽合成法Boc方法是经典的多肽固相合成法,以Boc作为氨基酸α-氨基的保护基,苄醇类作为侧链保护基,Boc的脱除通常采用三氟乙酸(TFA)进行。多肽合成时将已用Boc保护好的N-α-氨基酸共价交联到树脂上,TFA切除Boc保护基,N端用弱碱中和。肽链的延长通过二环己基碳二亚胺(DCC)活化、偶联进行,最终采用强酸氢氟酸(HF)法或三氟甲磺酸(TFMSA)将合成的目标多肽从树脂上解离。在Boc多肽合成法中,为了便于下一步的多肽合成,反复用酸进行脱保护,一些副反应被带入实验中,例如多肽容易从树脂上切除下来,氨基酸侧链在酸性条件不稳定等。2、Fmoc多肽合成法Carpino和Han以Boc多肽合成法为基础发展起来一种多肽固相合成的新方法——Fmoc多肽合成法。Fmoc多肽合成法以Fmoc作为氨基酸α-氨基的保护基。其优势为在酸性条件下是稳定的,不受TFA等试剂的影响,应用温和的碱处理可脱保护,所以侧链可用易于酸脱除的Boc保护基进行保护。肽段的最后切除可采用TFA/二氯甲烷(DCM)从树脂上定量完成,避免了采用强酸。同时,与Boc法相比,Fmoc法反应条件温和,副反应少,产率高,并且Fmoc基团本身具有特征性紫外吸收,易于监测控制反应的进行。Fmoc法在多肽固相合成领域应用越来越广泛。多肽液相分段合成随着多肽合成的发展,多肽液相分段合成(即多肽片段在溶液中依据其化学专一性或化学选择性,自发连接成长肽的合成方法)在多肽合成领域中的作用越来越突出。其特点在于可以用于长肽的合成,并且纯度高,易于纯化。多肽液相分段合成主要分为天然化学连接和施陶丁格连接。天然化学连接是多肽分段合成的基础方法,局限在于所合成的多肽必须含半光氨酸(Cys)残基,因而限定了天然化学连接方法的应用范围。天然化学连接方法的延伸包括化学区域选择连接、可除去辅助基连接、光敏感辅助基连接。施陶丁格连接方法是另一种基础的片段连接方法,其为多肽片段连接途径开拓了更广阔的思路。正交化学连接方法是施陶丁格连接方法的延伸,通过简化膦硫酯辅助基来提高片段间的缩合率。其他多肽合成方法1、氨基酸的羧内酸酐法(NCA)氨基酸的羧内酸酐的氨基保护基也可活化羧基。NCA的原理:在碱性条件下,氨基酸阴离子与NCA形成一个更稳定的氨基甲酸酯类离子,在酸化时该离子失去二氧化碳,生成二肽。生成的二肽又与其他的NCA结合,反复进行。NCA适用于短链肽片段的多肽合成,其周期短、操作简单、成本低、得到产物分子量高,在目前多肽合成中所占比例较大,技术也较为通用。2、组合化学法20世纪80年代,以固相多肽合成为基础提出了组合化学法,即氨基酸的构建单元通过组合的方式进行连接,合成出含有大量化合物的化学库,并从中筛选出具有某种理化性质或药理活性化合物的一套多肽合成策略和筛选方案。组合化学法的多肽合成策略主要包括:混合-均分法、迭代法、光控定位组合库法、茶叶袋法等。组合化学法的最大优点在于可同时合成多种化合物,并且能最大限度地筛选各种新化合物及其异构体。3、酶解法酶解法是用生物酶降解植物蛋白质和动物蛋白质,获得小分子多肽。酶解法因其多肽产量低、投资大、周期长、污染严重,未能实现工业化生产。酶解法获得的多肽能够保留蛋白质原有的营养价值,并且可以获得比原蛋白质更多的功能,更加绿色,更加健康。4、基因工程法基因工程法主要以DNA重组技术为基础,通过合适的DNA模板来控制多肽的序列合成。有研究者通过基因工程法获得了准弹性蛋白-聚缬氨酸-脯氨酸-甘氨酸-缬氨酸-甘氨酸肽(VPGVG)。利用基因工程技术生产的活性多肽还有肽类抗生素、干扰素类、白介素类、生长因子类、肿瘤坏死因子、人生长激素,血液中凝血因子、促红细胞生成素,组织非蛋白纤溶酶原等。基因工程法合成多肽具有表达定向性强,安全卫生,原料来源广泛和成本低等优点,但因存在高效表达,不易分离,产率低的问题,难以实现规模化生产。5、发酵法发酵法是从微生物代谢产物中获得多肽的方法。虽然发酵法的成本低,但其应用范围较窄,因为现在微生物能够独立合成的聚氨基酸只有ε-聚赖氨酸(ε-PL)、γ-聚谷氨酸(γ-PGA)和蓝细菌肽。[align=center][img=,770,348]https://ng1.17img.cn/bbsfiles/images/2019/03/201903151633244062_8177_3531468_3.jpg!w770x348.jpg[/img][/align]请移步百度搜“合肥国肽生物”即可我们主要提供:多肽合成、定制多肽、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。
Q1.引物是如何合成的?目前引物合成基本采用固相亚磷酰胺三酯法。DNA合成仪有很多种,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。(1) 去保护:加入Deblocking脱去碱基上5'- OH的保护基团DMT,获得游离的5'- OH;(2) 耦合:同时加入活化剂和新的碱基,新的碱基5'-OH仍然被DMT保护,3'端被活化与溶液中游离的5'-OH发生耦合反应;(3) 封闭:耦合反应中极少数5'- OH没有参加反应,用封闭试剂终止其后继续发生反应;(4) 氧化:加入氧化剂使其由核苷亚磷酸酯形成更稳定的核苷磷酸酯。Q2.引物合成后如何处理?切割与脱保护基:将合成好的寡核苷酸链从支持物上化学切割下来。常用新鲜的浓氨水来裂解CPG与初始核苷之间的酯键。断裂下来的寡核苷酸带有自由的3'羟基。纯化:根据所合成寡核苷酸的组成和应用来选定纯化的方法。常用的纯化方法有:C18、OPC、PAGE和HPLC。定量:根据寡核苷酸在260nm处的紫外吸收来定量。储存:分装抽干。Q3.需要什么级别的引物?根据实验需要,确定订购引物的纯度级别。应用引物长度要求纯度级别要求一般PCR扩增60 basePAGE诊断PCR扩增 40baseHEPD, PAGEDNA测序20base左右HEPD亚克隆,点突变等根据实验要求定HEPD, PAGE,HPLC基因构建(全基因合成)根据实验要求定HEPDPAGE反义核酸根据实验要求定HEPDPAGE修饰引物根据实验要求定PAGE, HPLCQ4.引物的质量是不是跟序列有关?四种碱基的性质和各自保护基的性质都有差别。所以合成难度是不一样的。难度最大的当属GC重复多的和序列中还有多个连续的G的引物。尤其对于后者,国内公司一般都做不了20个G以上的引物。实验证明,如果引物中有超过三个连续G的结构,传统方法得到的产物质量就会开始下降。而且目前通用的脱盐、OPC和PAGE方法都无效。鼎国昌盛生物公司拥有的HEPD专利技术能够克服高GC或者高G含量引物的合成和纯化障碍,对普通引物、高GC引物和无论长短的oligo d(G)都能得到同样的非常好的结果。Q5.需要合成多少OD数?根据实验目的确定。一般PCR扩增,2 OD引物,可以做500-1000次50ul标准PCR反应。如果是做基因拼接或退火后做连接,1 OD就足够了。Q6.如何计算引物的浓度?引物保存在高浓度的状况下比较稳定。一般情况下,我们建议将引物的浓度配制成100pmol/ul,称为保存浓度,而引物的工作浓度一般配制成10-50pmol/ul。加水的体积(微升)可直接参照合成报告单上推荐的体积,也可按下列方式计算:V (微升)= OD数x 33 x 10000 /引物的分子量引物的分子量可以从合成报告单上获得。注意:1 OD260= 33 ug/ml。Q7.如何计算引物的Tm值?引物设计软件都可以给出Tm,与引物长度、碱基组成及所使用缓冲夜的离子强度有关。长度为25base以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10 + 0.41 (GC%) - 600/size**公式中,Size =引物长度。Q8.引物(含修饰)的分子量是如何确定的?非修饰的引物的分子量(MW)在随引物提供的报告单上可以找到。如果需要估计一个引物的分子量按每个碱基的平均分子量为324.5,引物的分子量=碱基数 x碱基的平均分子量,或按下列公式计算MW= (NA * WA) + (NC * WC) + (NG * WG) + (NT * WT) +(Nmod * Wmod)+(Nx * Wx)+( NI* WI) +16* Ns-62.NA, NG, NC, NT, NI分别为引物中碱基A或G或C或T或I的数量,WA, WG ,WC, WT, WI分别为引物中碱基A或G或C或T或I的分子量,Nmod,Wmod分别为修饰基团的数目和分子量。对于混合碱基的分子量为混合碱基的分子量总合除以混合数,例如G+A混合的分子量为(313.21+329.21)/2 = 321.21。Ns为硫代数目,硫代每个位置增加分子量16。常规碱基分子量BaseMolecular WeightA313.21C289.18G329.21T304.19I314.2U290.17常规修饰基团分子量5’-Biotin405.453’-TAMARA623.605’-(6 FAM)537.463’-Dabsyl498.495’-HEX744.133’-(6 FAM)569.465’-TET675.243’-Amino Modifier C3153.075’-Cy5533.633’-Amino Modifier C7209.185’-Cy3507.593’-Thiol Modifier C3154.12Q9.如何溶解引物?干燥后的引物质地非常疏松,开盖前最好瞬时离心一下,或管垂直向上在桌面上敲敲,将引物粉末收集到管底。根据计算出的体积加入去离子无菌水或TE(pH8.0)缓冲液,室温放置几分钟,振荡助溶,离心将溶液收集到管底。溶解引物用的水一般不要用蒸馏水,因为有些蒸馏水的pH值比较低(pH4-5),引物在这种条件下不稳定。Q10.如何保存引物?引物合成后,经过一系列处理和纯化步骤,旋转干燥而成无色或白色絮状干粉。干 粉:运输时常温运输,-20℃可以保存一年。储存液:配好后分装成几管,避免反复冻融,-20℃可以保存半年。工作液:常规使用,4℃保存,也可-20℃保存,但应避免反复冻融。修饰荧光引物:需要避光保存,尽快使用为宜。Q11.最长可以合成多长的引物?我们合成过100base的引物,但是产率很低。除非需要,建议合成片段长度不要超过80base。引物越长,出现问题的概率就越大,按照目前的引物合成效率,大于80base的粗产品,全长引物的百分比不高,后续处理还有丢失很多,最后的产量很低。Q12.为什么修饰引物的产量要比一般引物低,价格要高?主要因为是修饰单体稳定性较差,偶连时间长,效率低,最后得到的产量自然低于一般的引物。修饰引物通常需要PAGE或HPLC纯化,纯化过程损失大。修饰引物使用的原料是一般引物原料的几百倍,所以产品的价格也高。Q13.引物片段退火后不能连接到载体上是什么问题?连接反应需要引物的5'磷酸基团。如果需要将合成的引物退火直接连接相应的载体上,引物需要磷酸化。磷酸化的产物如果还不能连接载体上,需要检查载体的酶切效果,需要改善引物退火的条件。SiRNA分子具有特殊的对称结构,退火的难度较大,退火时需要提高退火温度。Q14.为什么引物的OD260/OD280小于1.5?需
我们实验室做合成着色剂还在用聚酰胺粉涂板定性,近才听人家说有现成的展板,用完之后洗掉色素再活化就可以再用,想请用过的朋友介绍介绍下。

多肽合成方法分类多肽的合成主要分为两条途径:化学合成多肽和生物合成多肽。化学合成主要是以氨基酸与氨基酸之间缩合的形式来进行。在合成含有特定顺序的多肽时,由于多肽合成原料中含有官能度大于2的氨基酸单体,多肽合成时应将不需要反应的基团暂时保护起来,方可进行成肽反应,这样保证了多肽合成目标产物的定向性。多肽的化学合成又分为液相合成和固相合成。【合肥国肽生物】多肽液相合成主要分为逐步合成和片段组合两种策略。逐步合成简洁迅速,可用于各种生物活性多肽片段的合成。片段组合法主要包括天然化学连接和施陶丁格连接。近年,多肽液相片段合成法发展迅速,在多肽和蛋白质合成领域已取得了重大突破。在多肽片段合成法中,根据多肽片段的化学特定性或化学选择性,多肽片段能够自发进行连接,得到目标多肽。因为多肽片段含有的氨基酸残基相对较少,所以纯度较高,且易于纯化。多肽的生物合成方法主要包括发酵法、酶解法,随着生物工程技术的发展,以DNA重组技术为主导的基因工程法也被应用于多肽的合成。多肽的固相合成多肽的合成是氨基酸重复添加的过程,通常从C端向N端(氨基端)进行合成。多肽固相合成的原理是将目的肽的第一个氨基酸C端通过共价键与固相载体连接,再以该氨基酸N端为合成起点,经过脱去氨基保护基和过量的已活化的第二个氨基酸进行反应,接长肽链,重复操作,达到理想的合成肽链长度,最后将肽链从树脂上裂解下来,分离纯化,获得目标多肽。1、Boc多肽合成法Boc方法是经典的多肽固相合成法,以Boc作为氨基酸α-氨基的保护基,苄醇类作为侧链保护基,Boc的脱除通常采用三氟乙酸(TFA)进行。多肽合成时将已用Boc保护好的N-α-氨基酸共价交联到树脂上,TFA切除Boc保护基,N端用弱碱中和。肽链的延长通过二环己基碳二亚胺(DCC)活化、偶联进行,最终采用强酸氢氟酸(HF)法或三氟甲磺酸(TFMSA)将合成的目标多肽从树脂上解离。在Boc多肽合成法中,为了便于下一步的多肽合成,反复用酸进行脱保护,一些副反应被带入实验中,例如多肽容易从树脂上切除下来,氨基酸侧链在酸性条件不稳定等。2、Fmoc多肽合成法Carpino和Han以Boc多肽合成法为基础发展起来一种多肽固相合成的新方法——Fmoc多肽合成法。Fmoc多肽合成法以Fmoc作为氨基酸α-氨基的保护基。其优势为在酸性条件下是稳定的,不受TFA等试剂的影响,应用温和的碱处理可脱保护,所以侧链可用易于酸脱除的Boc保护基进行保护。肽段的最后切除可采用TFA/二氯甲烷(DCM)从树脂上定量完成,避免了采用强酸。同时,与Boc法相比,Fmoc法反应条件温和,副反应少,产率高,并且Fmoc基团本身具有特征性紫外吸收,易于监测控制反应的进行。Fmoc法在多肽固相合成领域应用越来越广泛。多肽液相分段合成随着多肽合成的发展,多肽液相分段合成(即多肽片段在溶液中依据其化学专一性或化学选择性,自发连接成长肽的合成方法)在多肽合成领域中的作用越来越突出。其特点在于可以用于长肽的合成,并且纯度高,易于纯化。多肽液相分段合成主要分为天然化学连接和施陶丁格连接。天然化学连接是多肽分段合成的基础方法,局限在于所合成的多肽必须含半光氨酸(Cys)残基,因而限定了天然化学连接方法的应用范围。天然化学连接方法的延伸包括化学区域选择连接、可除去辅助基连接、光敏感辅助基连接。施陶丁格连接方法是另一种基础的片段连接方法,其为多肽片段连接途径开拓了更广阔的思路。正交化学连接方法是施陶丁格连接方法的延伸,通过简化膦硫酯辅助基来提高片段间的缩合率。其他多肽合成方法1、氨基酸的羧内酸酐法(NCA)氨基酸的羧内酸酐的氨基保护基也可活化羧基。NCA的原理:在碱性条件下,氨基酸阴离子与NCA形成一个更稳定的氨基甲酸酯类离子,在酸化时该离子失去二氧化碳,生成二肽。生成的二肽又与其他的NCA结合,反复进行。NCA适用于短链肽片段的多肽合成,其周期短、操作简单、成本低、得到产物分子量高,在目前多肽合成中所占比例较大,技术也较为通用。2、组合化学法20世纪80年代,以固相多肽合成为基础提出了组合化学法,即氨基酸的构建单元通过组合的方式进行连接,合成出含有大量化合物的化学库,并从中筛选出具有某种理化性质或药理活性化合物的一套多肽合成策略和筛选方案。组合化学法的多肽合成策略主要包括:混合-均分法、迭代法、光控定位组合库法、茶叶袋法等。组合化学法的最大优点在于可同时合成多种化合物,并且能最大限度地筛选各种新化合物及其异构体。3、酶解法酶解法是用生物酶降解植物蛋白质和动物蛋白质,获得小分子多肽。酶解法因其多肽产量低、投资大、周期长、污染严重,未能实现工业化生产。酶解法获得的多肽能够保留蛋白质原有的营养价值,并且可以获得比原蛋白质更多的功能,更加绿色,更加健康。4、基因工程法基因工程法主要以DNA重组技术为基础,通过合适的DNA模板来控制多肽的序列合成。有研究者通过基因工程法获得了准弹性蛋白-聚缬氨酸-脯氨酸-甘氨酸-缬氨酸-甘氨酸肽(VPGVG)。利用基因工程技术生产的活性多肽还有肽类抗生素、干扰素类、白介素类、生长因子类、肿瘤坏死因子、人生长激素,血液中凝血因子、促红细胞生成素,组织非蛋白纤溶酶原等。基因工程法合成多肽具有表达定向性强,安全卫生,原料来源广泛和成本低等优点,但因存在高效表达,不易分离,产率低的问题,难以实现规模化生产。5、发酵法发酵法是从微生物代谢产物中获得多肽的方法。虽然发酵法的成本低,但其应用范围较窄,因为现在微生物能够独立合成的聚氨基酸只有ε-聚赖氨酸(ε-PL)、γ-聚谷氨酸(γ-PGA)和蓝细菌肽。[img=,457,333]https://ng1.17img.cn/bbsfiles/images/2019/04/201904221507346400_2482_3531468_3.jpg!w457x333.jpg[/img]我们主要提供:多肽合成、定制多肽、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。合肥国肽生物官网:http://www.bankpeptide.com欢迎咨询服务热线:17718122172;17718122684;17730030476;17718122397

10,抽取5个版友);中奖名单:m3071659(注册ID:m3071659)大川之子,纵横四海(注册ID:chuangu120)翠湖园(注册ID:hhx050)牛一牛(注册ID:v2700892)yifan1117(注册ID:yifan1117)http://ng1.17img.cn/bbsfiles/images/2017/03/201703141649_01_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703141649_02_708_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================可乐中合成着色剂的测定方法:SPE基质:可乐应用编号:103019化合物:柠檬黄 苋菜红 靛蓝 胭脂红 日落黄 亮蓝 赤藓红固定相:ProElut PLS色谱柱/前处理小柱:ProElut PLS 500mg/6mL 30/pkg样品前处理:样品准备/提取 取1 g可乐(加热超声驱除二氧化碳),40 μL甲酸于试管中,摇匀,作为上样液待净化。 SPE柱净化——ProElut PLS 500 mg/6 mL(Cat.# 68005) (1)活化/平衡:5 mL甲醇活化,5 mL水平衡,流出液弃去;(2)上样:将待净化液加入小柱,流出液弃去; (3)淋洗:5 mL 2%甲酸水溶液,流出液弃去,将小柱抽干; (4)洗脱:4 mL2%氨水-甲醇溶液洗脱,收集流出液,2%氨水-甲醇溶液调至中性; (5)重新溶解:在40 ℃下用减压蒸馏将收集液浓缩至近干,然后用50%甲醇水溶液定容至1 mL后供HPLC分析。色谱条件:HPLC应用101605文章出处:迪马科技应用实验室关键字:着色剂 钻石二代 可乐 柠檬黄 Diamonsil C18(2) 苋菜红 ProElut PLS 靛蓝 胭脂红 日落黄 亮蓝 赤藓红谱图:http://dikma.com.cn/Public/Uploads/images/kele2(1).GIF
在看有些SPE的应用文章时,发现在有的方法里用到好几种不同的SPE柱,有的SPE柱就专门强调在活化后不能抽干,而有的就没有强调,是不是没有专门强调的SPE柱就可以容许一定程度的抽干(筛板上无溶剂层)?
固相萃取柱的类型及应用 硅胶的填料(60A,40um)类型填料应用ODS(C18) 硅胶上键合十八烷基 反相萃取,适合于非极性到中等极性的化合物,比如,抗菌素, 巴比妥酸盐,酞嗪,咖啡因,药物,染料,芳香油,脂溶性维生素,杀 真菌剂,锄草剂,农药,碳水化合物,对羟基甲苯酸取代酯,苯酚, 邻苯二甲酸酯,类固醇,表面活化剂,茶碱,水溶性维生素。 Octyl (C8) 硅胶上键合辛烷 反相萃取,适合于非极性到中等极性的化合物,比如,抗菌素, 巴比妥酸盐,酞嗪,咖啡因,药物,染料,芳香油,脂溶性维生素,杀 真菌剂,锄草剂,农药,碳水化合物,对羟基甲基酸取代酯,苯酚,邻 苯二甲酸酯,类固醇,表面活化剂,水溶性维生素。 Ethyl (C2)硅胶上键合乙基 相对C18和C8,因为短链,保持作用小的多,适合非极性化合物。 Phenyl 硅胶上键合苯基 相对C18和C8,反相萃取,适合于非极性到中等极性的化合物 Silica 无键合硅胶 极性化合物萃取,如乙醇,醛,胺,药物,染料,锄草剂,农药,酮,含氮类化合物,有机酸,苯酚,类固醇 Cyano(CN) 硅胶上键合丙氰基烷 反相萃取,适合于中等极性的化合物,正相萃取,适合于极性 化合物,比如,黄曲霉毒素,抗菌素,染料,锄草剂,农药,苯酚,类 固醇。弱阳离子交换萃取,适合于碳水化合物和阳离子化合物。 Amino(NH2) 硅胶上键合丙氨基 正相萃取,适合于极性化合物。弱阴离子交换萃取,适合于 碳水化合物,弱性阴离子和有机酸化合物。 Strong Anion Exchange(SAX) 硅胶上键合卤化季氨盐 强阴离子交换萃取,适合于阴离子,有机酸,核酸,核苷酸, 表面活化剂。容量:0.2毫当量/克。 Strong Cation Exchange(SCX) 硅胶上键合磺酸钠盐 强阳离子交换萃取,适合于阳离子,抗菌素,药物,有机碱,氨基酸,儿茶酚胺,锄草剂,核酸碱,核苷,表面活化剂。容量:0.2毫 当量/克。 AL2O3填料 类型填料应用Alumina A(acidic)酸性 PH ~5 极性化合物离子交换和吸附萃取,如维生素. Alumina B(basic) 碱性 PH~8.5 吸附萃取和阳离子交换。 Alumina N(neutral) 中性 PH~6.5 极性化合物吸附萃取。调节pH,阳和阴离。 子交换.适合于维生素,抗菌素,芳香油,酶,糖苷,激素 Florisil填料-硅酸镁 类型填料应用Florisil 极性化合物的吸附萃取,如乙醇,醛,胺,药物,染料,锄草剂,农药,PCBs,酣,含氮类化合物,有机酸,苯酚,类固醇 类型填料应用EVIDEXII(Drugs of Abuse辛烷和阳离子交换树脂 Amphetamina/Methamphetamine、 PCP、Benzoylecgonine、 Codeine/Morphine、 THC-COOH(Marijuana)
固相萃取柱的类型及应用类型 填料 应用 硅胶的填料(60A,40um)ODS(C18) 硅胶上键合十八烷基 反相萃取,适合于非极性到中等极性的化合物,比如,抗菌素, 巴比妥酸盐,酞嗪,咖啡因,药物,染料,芳香油,脂溶性维生素,杀 真菌剂,锄草剂,农药,碳水化合物,对羟基甲苯酸取代酯,苯酚, 邻苯二甲酸酯,类固醇,表面活化剂,茶碱,水溶性维生素。 Octyl (C8) 硅胶上键合辛烷 反相萃取,适合于非极性到中等极性的化合物,比如,抗 菌素, 巴比妥酸盐,酞嗪,咖啡因,药物,染料,芳香油,脂溶性维生素,杀 真菌剂,锄草剂,农药,碳水化合物,对羟基甲酸取代酯,苯酚,邻 苯二甲酸酯,类固醇,表面活化剂,水溶性维生素。 Ethyl (C2) 硅胶上键合乙基 相对C18和C8,因为短链,保持作用小的多,适合非极性化合物。 Phenyl 硅胶上键合苯基 相对C18和C8,反相萃取,适合于非极性到中等极性的化合 物, Silica 无键合硅胶 极性化合物萃取,如乙醇,醛,胺,药物,染料,锄草剂,农 药,酮,含氮类化合物,有机酸,苯酚,类固醇 Cyano(CN) 硅胶上键合丙氰基烷 反相萃取,适合于中等极性的化合物,正相萃取,适合于极性 化合物,比如,黄曲霉毒素,抗菌素,染料,锄草剂,农药,苯酚,类 固醇。弱阳离子交换萃取,适合于碳水化合物和阳离子化合物。 Amino(NH2) 硅胶上键合丙氨基 正相萃取,适合于极性化合物。弱阴离子交换萃取,适合于 碳水化合物,弱性阴离子和有机酸化合物。 Strong Anion Exchange(SAX) 硅胶上键合卤化季氨盐 强阴离子交换萃取,适合于阴离子,有机酸,核酸,核苷酸, 表面活化剂。容量:0.2毫当量/克。 Strong Cation Exchange(SCX) 硅胶上键合磺酸钠盐 强阳离子交换萃取,适合于阳离子,抗菌素,药物,有机碱,氨基酸,儿茶酚胺,锄草剂,核酸碱,核苷,表面活化剂。 容量:0.2毫 当量/克。 AL2O3填料 Alumina A(acidic) 酸性 PH ~5 极性化合物离子交换和吸附萃取,如维生素. Alumina B(basic) 碱性 PH~8.5 吸附萃取和阳离子交换。 Alumina N(neutral) 中性 PH~6.5 极性化合物吸附萃取。调节pH,阳和阴离。子交换.适合于维生素,抗菌素,芳香油,酶,糖苷,激素 Florisil填料-硅酸镁 Florisil 极性化合物的吸附萃取,如乙醇,醛,胺,药物,染料,锄草 剂,农药,PCBs,酣,含氮类化合物,有机酸,苯酚,类固醇 用于NIDA-5所要求的五种毒 品代谢物分析EVIDEXII(Drugs of Abuse) 辛烷和阳离子交换树脂 Amphetamina/Methamphetamine、 PCP、Benzoylecgonine、 Codeine/Morphine、 THC- COOH(Marijuana)
选用的阳离子SCX固相萃取小柱,测肌肉中的卡巴多,和喹乙醇很像,是一种促生长剂,可使用柱子的时候,回收率很低,请问哪位大侠用过这种柱子啊,我查了资料,使用5ML甲醇,5ML水,5ML稀盐酸活化,可样品液过柱损失不小啊,影响回收率,请问前对样品液的PH值要求高吗,还有淋洗和洗脱选用什么比较合适啊,我试了用水和甲醇淋洗,都有损失,郁闷啊
liner在去活化之前应先用溶剂或肥皂水将其刷洗干净, 另外可以用木质柄的棉花棒, 于溶剂或肥皂水中刷洗liner的玻璃管内部, 如无木质柄的棉花棒,则一般的塑料柄的棉花棒则只可于肥皂水中刷洗, 最后冲洗干净,干燥后进行去活化的步骤. 干燥后的liner先以玻璃吸管吸取甲醇冲洗, 反复数次, 然后溶剂换成EA,以玻璃吸管吸取冲洗liner, 最后把溶剂换成n-hexane重复前面所述的步骤, 要注意的是溶剂使用顺序不可以变动!!将二氯二甲基硅烷和甲苯以1:9的体积混合,并以表玻璃加盖 ,需注意盛装这个混合液的玻璃容器,本身也会与前述的混合液反应, 故在使用后会有一段时间其容器内部会呈现疏水性质, 将前面以n-hexane洗干净的liner放入其中,要注意在liner的管壁上绝对不可以出现气泡, 当所有的liner都放好后,盖上表玻璃, 于抽气厨中静置1~2小时 !!将反应完成之liner取出(每次一支, 其余的浸泡在反应剂中,绝对不要一次将所有的liner拿出来暴露于空气中), 先以玻璃吸管吸取n-hexane冲洗, 然后以EA冲洗, 最后是以甲醇冲洗,顺序不可以颠倒!! n-hexane是要洗掉未反应的二氯二甲基硅烷, 而最后的甲醇是要和在已经和玻璃表面反应的二氯二甲基硅烷, 另外的一个-Cl基团反应(end cap), 所以如果顺序搞错的话是很凄惨的事情。上面的步骤结束后,可以将liner置于高温烘箱中,在300度下烘一个晚上!! 隔天早上取出时, 须于干燥皿或烘箱中降温, 而不要于空气中, 这样你的liner会很快速的开始吸收水气!最后将liner置放于防潮箱中保存!!反应的副产物是气态HCl, 要全程在抽气橱中操作!!! 二氯二甲基硅烷会与空气中的水分反应, 注意存放的条件!!
中国科技网讯 据《自然》杂志网站8月5日(北京时间)报道,韩国研究人员发现,一种新的合成分子有望成为治疗结核病的候选药物,小鼠实验已经证实了其疗效:该合成分子可以抑制结核杆菌生长,同时,与现有的很多抗结核药物相比,细菌更难以对其产生耐药性。如果临床试验证明其对人类安全、有效,将有望挽救更多人的生命。 韩国巴斯德研究所微生物学家凯文·派特领导的一个研究小组耗时5年,调查了超过12万种化合物。他们用结核分枝杆菌感染小鼠体内被称为巨噬细胞的免疫细胞,然后观察哪些化合物能够抑制细菌生长,最终从中筛选出了一种合成分子进行深入评估。 研究人员在《自然·医学》杂志上报告称,这种合成抗菌分子具有新的作用机制——抑制ATP(一种为细胞的大多数酶提供能量的化合物)的合成,从而阻止结核分枝杆菌生长。他们开展的测试表明,该合成分子能够成功治疗小鼠结核病。 南非开普敦大学结核病方面的生物学家瓦莱丽·米兹拉希说,这项研究“肯定了一个观念,即有新的结核病药物靶标在等待着被发现,方法就是筛选不同的合成分子库”。 但这个阶段的成功并不能保证该合成分子可用于有效地治疗人类肺结核。派特说,明年将在一小群健康志愿者身上进行该候选药物的一期临床试验,以评估其安全性和耐受性。不过,就制药业的现状而言,即使进入一期临床试验,也只有5%的药物最终能获准上市销售。 如果这种药物最终进入临床应用,另一个挑战将是防止结核杆菌快速进化出“对策”。但研究人员表示,该分子属于一类新的合成化学物质,与现有的抗结核药物没有相似之处,这可能使得细菌难以对它产生耐药性,从而无法发展出耐药菌株。 派特和他的团队计划继续寻找更多的候选抗结核药物分子。结核病通常需要采用“鸡尾酒”疗法,派特希望他们的研究能为临床医生提供更多不同药物。(记者 陈丹) 总编辑圈点 从林妹妹到茶花女,很多文学巨匠笔下,都有一位面色白皙却两颊绯红,多愁善感而楚楚动人的女主角,给她们带来这种共同特征的正是结核病。文学作品中的“性感”和“美丽”并不是美化,相反证明了那时候人们对这一充满神秘感的不治之症的畏惧。时至今日,结核病依然在侵蚀很多人的健康,其中的超级耐药结核病更是难缠。新的合成药物具有新的作用机制,尤其是在抗耐药方面的不俗表现,使它有可能成为征服这种古老疾病的重要武器。另外,对“合成”的思路加以应用,也可能在对抗其他耐药病菌上带来惊喜。 《科技日报》(2013-8-6 一版)
【序号】:1【作者】:申艳红 张文升 刘启宾 渠桂荣 【题名】:环磷腺苷的合成【期刊】:《中国医药工业杂志》 【年、卷、期、起止页码】:2004年03期【全文链接】:http://www.cnki.com.cn/Article/CJFDTotal-ZHOU200403001.htm【序号】:2【作者】:张今 刘兰英 沈雨生 【题名】:3′,5′-环化腺苷酸的合成【期刊】:《吉林大学学报(理学版)》 【年、卷、期、起止页码】:1979年02期 【全文链接】: http://www.cnki.com.cn/Article/CJFDTotal-JLDX197902008.htm 【序号】:3【作者】:张今 刘兰英 沈雨生 【题名】: 3′,5′-环化腺苷酸合成的改进【期刊】:《中国医药工业杂志》 【年、卷、期、起止页码】:1979年04期 【全文链接】:http://www.cnki.com.cn/Article/CJFDTotal-ZHOU197904002.htm谢谢!谢谢!
食品中人工合成着色剂的测定解决方案合成着色剂因其色彩亮丽、性质稳定、价格低廉,成为食品工业常用的添加剂之一。它们通常是以苯、甲苯、萘等化工原料合成,长期食用对人体健康具有一定的毒性,尤其是对少年儿童。世界各国对合成着色剂的使用范围和限量都有严格的规定,《GB 2760-2014 食品安全国家标准 食品添加剂使用标准》中准许使用的人工合成色素也有严格规定。我国也有很多检测着色剂的相关标准,采用不同的净化方法,包括C18柱净化、聚酰胺粉净化等,C18净化方法仅仅适用于水果罐头,而聚酰胺粉净化方法过程比较繁琐。方法优势:对比国标方法本方案具有:前处理步骤简单、回收率高、方法稳定性好、净化效果优异等特点;采用了迪马科技的ProElut PWA-2,可以保证实验结果的重现性和准确性;过柱方法简单易操作,对操作人员要求不高,检测成本相对较低,能被很多企事业单位采用;可同时检测8种人工合成着色剂,检出限:亮蓝为1.0 mg/kg,其他均为0.2 mg/kg,符合国家标准要求。专用柱优势ProElut PWA-2吸附剂采用弱阴离子交换机理去除杂质,同时对着色剂没有不可逆的吸附,保证了样品的净化效果及回收率;本产品是商品化的成品柱,吸附剂稳定性好,不受外界环境因素影响,保证实验结果的重现性和准确性;过柱过程操作步骤简单,节省时间,提高了工作效率。以下为详细解决方案,敬请参考!食品中人工合成着色剂的测定1、适用范围本方案适用于糕点、果酱、水果罐头、黑芝麻糊、果冻、冰淇淋、乳饮料、糖果和红酒中人工合成着色剂的检测;检出限:亮蓝为1.0 mg/kg,其余均为0.2 mg/kg;2、提取2.1 糕点、果酱取1.0 g样品,加入20 mL提取液A*,振荡2 min,40 ℃水浴超声提取10 min,6000 rpm下离心2 min,收集上清液;取下层残留物,加入10 mL提取液A*,振荡2 min,40 ℃水浴超声提取15 min,6000 rpm下离心2 min,收集上清液;将下层残留物用10 mL提取液A* 按照步骤(2)重复提取一次,合并三次上清液;将上清液在40 ℃水浴条件下,减压蒸至约15 mL,再加入3 mL甲酸混匀,待净化。2.2 水果罐头、黑芝麻糊取1.0 g样品,加入20 mL提取液A*,振荡2 min,40 ℃水浴超声提取10 min,6000 rpm下离心2 min,收集上清液;取下层残留物,加入10 mL提取液A*,振荡2 min,40 ℃水浴超声提取15 min,6000 rpm下离心2 min,合并两次上清液;将上清液在40℃水浴条件下,减压蒸至约15 mL,再加入3 mL甲酸混匀,待净化。2.3 冰淇淋取1.0 g样品,加入20 mL提取液A*,振荡2 min,40 ℃水浴超声提取10 min,6000 rpm下离心2 min,收集上清液;将上清液在40 ℃水浴条件下,减压蒸至约15 mL,再加入3 mL甲酸混匀,待净化。2.4 果冻取1.0 g样品,加入10 mL水,40 ℃水浴超声提取15 min,加入5 mL甲醇和3 mL甲酸混匀,待净化。2.5 乳饮料、糖果取1.0 g样品,加入10 mL水、5 mL甲醇和3 mL甲酸混匀,待净化。2.6 红酒取1.0 mL样品,加入0.5 mL甲醇和0.3 mL甲酸混匀,待净化。*提取液A:取100 mL乙醇和50 mL乙腈,混匀,取140 mL乙醇:乙腈(2:1)混合溶液,加入60 mL水和2 mL氨水,混匀。3、净化——ProElut PWA-2 150 mg/6 mL(Cat.# 65815)a活 化:依次用5 mL甲醇、5 mL10%甲酸水活化;b上 样:加入待净化液,弃去流出液;c淋 洗:加入5 mL甲醇,弃去流出液;d洗 脱:加入5 mL 15%氨水甲醇溶液,收集流出液;e重新溶解:将洗脱液在50 ℃下氮吹至约300 μL,用流动相定容至1 mL,供HPLC分析。4、色谱条件色谱柱:Inspire C18,250 × 4.6 mm,5 μm(Cat.# 81006)流 速:1.0 mL/min进样量:20 μL柱 温:35 ℃检测器:PDA 254 nm流动相:A:乙腈 B:0.02 mol/L 乙酸铵溶液梯度设置时间(min)020303140A(%)5303755B(%)95706395955、添加回收结果 柠檬黄新红苋菜红靓蓝日落黄诱惑红亮兰赤藓红糕点97.4595.4792.8092.7097.6999.7787.8091.38果酱96.7995.3194.2194.0795.2398.3693.9192.55果冻107.12101.29103.2996.04103.18103.96100.3693.87冰淇淋99.5597.66106.9893.44101.45102.22[
各位老师好,我单位用802E型饱和甘汞电极和氟离子电机组合测定水氟,在使用前电极厂家建议在纯水中浸泡4小时就可以,但单位老同事说饱和甘汞电极不可以泡在纯水中,那应该在什么溶液中活化?
1、清洗剂的残余物导致不正确的分析结果2、高度敏感的分析方法要求彻底清洁实验室玻璃器皿3、残余物可能带来交叉污染4、来自清洗过程的表面活化剂的残余物影响细菌和细胞培养的生长5、残余物可能会产生挥发性的有害物质。
我想和大家讨论一个关于草甘瞵和草甘瞵铵盐的问题,我公司用草甘瞵合成草甘瞵铵盐,我用液相色谱分析草甘瞵铵盐,但是发现样品溶解性不好,我觉得可能是合成不完全,还有一部分草甘瞵在里面,因为色谱分析只有草甘瞵出峰,铵盐使用分析出来的草甘瞵的含量乘以1.1005后得出的结果,所以导致乘以系数后含量到了100%以上,是不是草甘瞵如果反应彻底的话,应该很好溶解才对,也就是草甘瞵铵盐很好溶解,如果有不溶物,说明肯定有草甘瞵存在,对不对?还有像这种情况用什么方法才能将里面的草甘瞵和草甘瞵铵盐的含量分别检测出来呢?用液谱好象不行吧,因为液谱铵盐是不出峰的,有没有化学方法可以检测出来,也就是说看一看合成效果怎么样?

10,抽取5个版友);中奖名单:大川之子,纵横四海(注册ID:chuangu120)zengzhengce163(注册ID:zengzhengce163)翠湖园(注册ID:hhx050)sixingxing(注册ID:v2889187)夏天的雪(注册ID:bingwang228)http://ng1.17img.cn/bbsfiles/images/2017/03/201703081529_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703081529_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================可乐中合成着色剂的测定方法:SPE基质:非酒精饮料应用编号:103019化合物:柠檬黄 苋菜红 靛蓝 胭脂红 日落黄 亮蓝 赤藓红固定相:ProElut PLS色谱柱/前处理小柱:ProElut PLS 500mg/6mL 30/pkg样品前处理:样品准备/提取取1 g可乐(加热超声驱除二氧化碳),40 μL甲酸于试管中,摇匀,作为上样液待净化。SPE柱净化——ProElut PLS 500 mg/6 mL(Cat.# 68005)(1)活化/平衡:5 mL甲醇活化,5 mL水平衡,流出液弃去;(2)上样:将待净化液加入小柱,流出液弃去;(3)淋洗:5 mL 2%甲酸水溶液,流出液弃去,将小柱抽干;(4)洗脱:4 mL2%氨水-甲醇溶液洗脱,收集流出液,2%氨水-甲醇溶液调至中性;(5)重新溶解:在40 ℃下用减压蒸馏将收集液浓缩至近干,然后用50%甲醇水溶液定容至1 mL后供HPLC分析。色谱条件:色谱柱:Diamonsil C18(2) 250×4.6 mm ID,5 μm (Cat. #99603) 流 速:1.0 mL/min 检测器:UV 254 nm 柱 温:35 ℃ 进样量:20 μL 流动相A:0.05 mol/L乙酸铵 流动相B:乙腈 T(min) 0(A 95%);20(A 70%);30(A 63%);31(A 95%);35(A 95%)文章出处:迪马科技应用实验室关键字:着色剂 钻石二代 可乐 柠檬黄 Diamonsil C18(2) 苋菜红 ProElut PLS 靛蓝 胭脂红 日落黄 亮蓝 赤藓红谱图:http://dikma.com.cn/Public/Uploads/images/kele2(1).GIF
吊白块又称雕白块,是一种白色块状或结晶性粉粒的有机化合物,化学名为甲醛次硫酸氢钠,分子式为NaHSO2CH2O2H2O,溶于水,在常温下较为稳定,在高温时分解成亚硫酸盐,有强还原性。是由锌粉和二氧化硫反应生成低亚硫酸锌,再与甲醛和锌粉作用后,在真空蒸发浓缩、凝结成块而制得。反应物中通常含有亚硫酸氢钠、甲醛等。 吊白块是一种工业用漂白剂,印染丝绸用的工业原料,也可用作洗涤剂和除垢剂的原料,主要应用于印染工业如棉布、人造丝、天然纤维、织布等拔染剂,及用作丁苯橡胶和合成树脂活化剂等。 吊白块是国家禁止在食品中使用的有害物质,卫生部门曾多次发文明令禁止,但不法分子利用吊白块可以使食品增白使食品外观色泽亮丽,能延长食品保存时间可以防腐,能增加食品的韧性,使食品久煮不糊,吃起来爽口等特点,在利益的驱动下,置人民的健康不顾,屡禁屡用。食品中掺入吊白块后,会破坏食品的营养成分,食用后引起人体过敏、刺激肠道、食物中毒等疾患,严重者影响视力,甚至可能致癌。在加工过程中分解产生的甲醛,是细胞原浆毒,能使蛋白质凝固,对人体的肝脏、肾脏等有严重损害,长期服用,对中枢神经系统也会造成累积性损害,导致失眠,生物节律紊乱,引起四肢麻木或震颤,影响人体的代谢机能,一次食用剂量达到10g的,会有生命危险。
请问大家植物中的草甘磷、草胺磷检测有什么关键点,2g样品加20ml水提取,20ml二氯甲烷除酯,用5ml甲醇、5ml水活化,取提取液5ml过阴离子交换柱,得到样品基质,拿这个去配标,然后用500ul样品基质+300ul1%芴甲氧羰酰氯+200ul5%硼酸钠,40度衍生后,上[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]质,出一堆峰,进了一个梯度,没有线性,找不到目标峰。优化找的离子对404.1/136/179、392.1/88/214,大家有啥好的方法提供参考?







 我要推广仪器
我要推广仪器
 下载APP
下载APP