[em0815] 哪为老师有盐酸多西环素和多西环素一水物标准BP2008和EP6.0版部分.请分享一下.不胜感激.急需.
我在称量一水戊烷磺酸钠时,天平的数字一直在往上加是为什么呢?一直加都不停止。是不是戊烷磺酸钠会吸水呢
请问各位:在哪可买到化学纯或更好的光谱纯一水硫酸亚铁?知道的请告诉下,谢谢!
我们公司买了一水乙酸钙,用卡氏滴定法测得水分含量为百分之八左右,但从分子量上来看,一个结晶水的质量百分比都有百分之十,请教大侠这是为什么?卡费休试剂能与一水乙酸钙中的结晶水反应吗
乳糖为一水化合物,做出来的红外图谱与图谱集有出入,请问乳糖是否要干燥后再做红外?怎样干燥呢?其检测方法上无干燥失重和水分项目的检测,那我应该在多少度干燥呢?谢谢了~!
哪位大侠有英国药典98/93版?急需关于一水柠檬酸的测试方法。多谢分享!
来了一批饲料级一水硫酸锌完全按标准方法,滴定终点难判定,近终点时后继续滴定,溶液一直维持为浅红色,滴定过量后,放置大约5分钟后,溶液转为亮黄色,做对比试验,取以前检测合格的一水硫酸锌平行操作,结果终点明显。怀疑新样品中存在干扰金属离子与指示剂和EDTA的络合稳定常数接近,导致变色缓慢,但不知道什么离子会的这种现象,大家有没有类似的经历,帮我分析一下原因。
如题,食用的葡萄糖是一水的还是无水的呢?
[font=SimSun, STSong, &]今天接到举报,辖区内有人在自家使用工业醋酸勾兑食用醋。被举报人家中设有食用醋生产作坊基本设施,但在被举报人家中发现“一水柠檬酸”一袋。[/font][font=SimSun, STSong, &] 被举报人是使用高粱制作食用醋,是粮食醋。[/font][font=SimSun, STSong, &] 我在GB2760中没有查到关于一水柠檬酸的信息,但是在网上查到有人说,柠檬酸和一水柠檬酸没什么区别,这种说法对吗?[/font][font=SimSun, STSong, &] 另外,醋中是否允许添加一水柠檬酸?可以的话有没有限量要求?[/font][font=SimSun, STSong, &] 我们打算对被举报人生产的醋做一次抽检,需要注意什么?如果被举报人的醋是勾兑的,需要检测什么项目?[/font]
请问哪位高人知道工业一水硫酸锌中硫酸根含量的测定方法~?不胜感谢!
一水葡萄糖中金属元素的检测,或者一些国标
请问如何测定一水软铝石的比表面?谢谢. 工作需要,请各位不吝指教.
求教各位:做葡萄糖标准曲线时,是否能用一水葡萄糖?一水葡萄糖在105摄氏度恒重后,是否能除去结晶水??谢谢!!!
[color=#0021b0]急需BP98关于一水柠檬酸的要求和测试方法,哪位大侠有,麻烦帮忙解个围,谢谢![img]http://simg.instrument.com.cn/bbs/images/brow/em09511.gif[/img][/color]
谁有德国生产的超纯水仪(TKA)的使用说明书?谢谢.
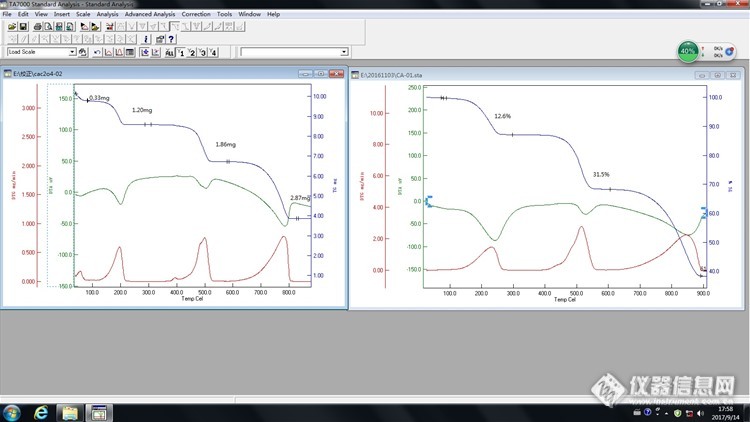
[img=,690,295]http://ng1.17img.cn/bbsfiles/images/2017/09/201709150830_01_3223531_3.jpg[/img]实验室有一台热重差热综合热分析仪,我买了些一水草酸钙的样品,今天测的时候有些异常现象,图里左边是我做的,右边的是厂家安装时做的。 第一个问题是试样放好后室温下样品质量就一直较匀速的下降,曲线上开始部分也能看见质量下降了0.33mg,对应DTA有个小吸热峰。然后后面的失重就偏差比较大了。一水草酸钙100℃以下也不会分解,难道是不够干燥? 第二个问题是400度左右出现一个小的放热峰,DTG也有峰。做了两次实验都出现这情况,难道是样品里有其他的杂质?
建筑工地有一处渗水,附近恰有一处管道漏水,如何快速判定这两处污水是否为同一水源呢?如要做水质分析,应该测哪些项目呢?没有一点头绪,急求助!先谢过了!
[size=2]请问:我的目标物既可溶于水,又可溶于甲醇乙腈等有机溶剂,目前样品溶液是水+甲醇体系,我如何将其浓缩,例如从3mL到1mL。我知道可以[b]旋蒸[/b],还有没有其他的好办法呢?因为水的比例较多,加无水硫酸钠可怕不是很合适!:(谢谢![/size]
[align=center]~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~[/align] 我想请问各位朋友,用GC-FID测同一水相中的氯己烷、二氯甲烷、三氯甲烷、四氯化碳各自的浓度,可行吗?有没有哪位朋友测过呢?检出限大致的多少,急求
AKATA制备型液相色谱蛋白分析仪纯化蛋白步骤很简单的,比较适合初学者。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=113913]AKATA制备型液相色谱蛋白分析仪纯化蛋白步骤[/url]
问题:根据《危废管理名录》2021年版规定,请问水性聚乙烯醇胶水是否属于危险废物,水性聚乙烯醇胶水的污泥和包装材料又是否属于危废?回复:您好,《国家危险废物名录》编制组针对《名录》修订情况及使用过程中常见的一些问题编写的解答材料提及,“不包括水性漆”是指水性漆渣不属于列入《名录》的危险废物,其是否属于危险废物需要根据《危险废物鉴别标准》(GB5085.1~7)《危险废物鉴别技术规范》(HJ 298)等予以判定。链接详见http://www.craes.cn/glzc/202012/t20201223_814505.shtml。有关《国家危险废物名录》解读释义事宜,需咨询编制发布等有法定解释权的单位。感谢您的关注与支持!
Millipore纯水机中的SD卡中存有几M软件,是纯水机的启动应用软件,很容易损坏。一旦过保损坏,厂家修复报价¥4000。备份方法:1.切断电源。2.打开右侧后盖。3.向里推SD卡,即自动弹出。4.用读卡器读出全部文件,拷贝-保存(最好压缩)。
为什么超纯水要即刻使用而不宜储存在工作中,我们常被问到下列的问题:“为什么超纯水制造机所显示的阻抗值明明是18.2MΩ.cm,但实验结果还是怪怪的 !!”“因为18.2MΩ.cm是超纯水的水质指标,所以可否将之做为更换耗材的唯一依据,而不需要考虑时间因素” 。 首先,我们需要先说明一下超纯水的特性!我想,大家都会同意”水”是超机溶剂,所以即使是自来水中也会含有科学实验所不能忍受的千万种杂质,因此我们都会用最先进的技术来纯化水质,而造成一种极度人工与环境极不平衡的水,它的名字叫做”超纯水”,当这种水从纯水系统制造出来的瞬间,即刻开始与其接触的环境产生溶解反应,我们戏称这种水为 “ hungry water” , 它会从空气中吸收杂质,如悬浮粉尘,挥发性有机物VOC以及微生物等,它也会从容器中吸收化学溶出物来,包含有机或无机物在ppb的层级上。还有,它又与空气中的二氧化碳发生变化,对已经纯化成超纯水的水而言,二氧化碳→碳酸所带来的酸硷变化就非常有趣了。首先,空气中二氧化碳的浓度虽然不高,只有0.038%(380ppm),却能与水产生化学反应如下:CO2(g)+H2O(l) «H2CO3(l)碳酸是一种弱酸(Ka1=4.3×10-7),但由于超纯水中已无任何主导性(dominant)的相对强酸,强碱,共轭酸,共轭硷的情况下,碳酸是唯一主导性性的弱酸,也是唯一离子的来源(请忽略掉H2O的解离)。Ka1= / =4.3×10-7如有需要,任何时间或地点,我们都可以模拟出二氧化碳→碳酸→碳酸根离子的现象,当超纯水开始曝露在大气下时,二氧化碳的溶解,就会无可避免的持续下去,这时,我们可以用电导率(conductivity)或pH的变化来监视这个过程(请参考附录1).因为水中的离子浓度持续增加,所以电导率会持续升高(或电阻抗值持续下降),通常[
化妆品中卡波姆的分离提取分析目标对象是化妆品中的卡波姆的分离提取。卡波姆:丙烯酸与烯丙基蔗糖交联的高分子聚合物,在化妆品中作为增稠剂,粘度大,在水、乙醇、甲醇中先溶胀,查资料说溶于水、乙醇中,但60度下加热搅拌了20min还没有完全溶解,不知是否还需要更长时间?不溶于乙酸乙酯、石油醚,但是在乙醇溶液中加了乙酸乙酯(作为沉淀剂)后也没有看到沉淀析出。不知该如何进行下去了,请有经验的高手指点指点,不甚感激!
一. EDTA 的离解平衡 在水溶液中, 2 个羧基 H + 转移到氨基 N 上,形成双极离子: EDTA 常用 H 4 Y 表示,由于其在水及酸中的溶解度很小,常用的为其二钠盐: Na 2 H 2 Y 2H 2 O ,也简写为 EDTA 。 当溶液的酸度很高时,两个羧基可再接受 H + ,形成 H 6 Y 2+ ,相当一. EDTA的离解平衡在水溶液中,2个羧基 H+转移到氨基N上,形成双极离子: http://www.foodmate.net/jianyan/lihua/fenxi/5.files/image001.gif EDTA 常用 H4Y 表示,由于其在水及酸中的溶解度很小,常用的为其二钠盐:Na2H2Y·2H2O ,也简写为EDTA 。 当溶液的酸度很高时,两个羧基可再接受H+ ,形成H6Y2+ ,相当于一个六元酸,有六级离解常数: Ka1=10-0.9 Ka2=10-1.6 Ka3=10-2.1 Ka4=10-2.8 Ka5=10-6.2 Ka6=10-10.3 七种形式: H6Y2+ 、H5Y+ 、H4Y 、H3Y- 、H2Y2- 、HY3- 、Y4- 当 pH 11 时,主要以 Y4- 形式存在——配位离子二. M-EDTA 的特点1. EDTA具有广泛的配位性能,几乎能与所有的金属离子形成稳定的螯合物 有利之处:提供了广泛测定元素的可能性(优于酸碱、沉淀法) 不利之处:多种组分之间易干扰——选择性2. EDTA与形成的M- EDTA 配位比绝大多数为1:13. 螯合物大多数带电荷,故能溶于水,反应迅速三. EDTA配合物的配位平衡及其影响因素(一) EDTA配合物的稳定常数 为简便,金属离子与EDTA的反应常将电荷略去写成通式: 配位平衡 M + Y == MY在配位滴定过程中,当溶液中没有副反应发生时,当反应达平衡时,用绝对稳定常数 KMY 衡量配位反应进行的程度: 稳定常数 http://www.foodmate.net/jianyan/lihua/fenxi/5.files/image003.gif (KMY 越大,配合物越稳定) (1)(KMY 不因浓度、酸度及其它配位剂或干扰离子的存在等外界条件而改变)
请教各位专家,怎样分离 乙醇:水:乙酸异丁酯的混合物,比例约为10:5:85,目标是尽量减少乙醇和水,谢谢!
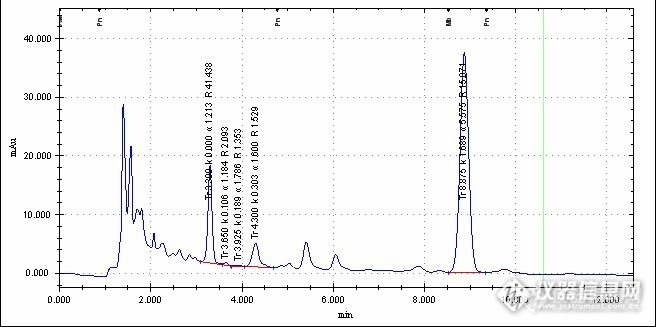
锁阳咖啡中咖啡因含量的测定锁阳咖啡以天然锁阳为基础,配以速溶咖啡,是近几年涌现出的一种新型的固体饮料。锁阳主产于甘肃、青海、内蒙,其中以甘肃嘉酒地区的锁阳产量最大、质地最优。锁阳可调节生理机能、均衡营养、促进血液循环、滋肝健肾。锁阳咖啡是锁阳和咖啡的有机结合,越来越受到消费者的青睐。但是人们在饮用锁阳咖啡的同时却容易忽略咖啡的品质,咖啡因是咖啡中的一种重要成分,也是衡量其质量的一项重要指标,作为一种中枢兴奋剂,能兴奋大脑皮层,但易上瘾,因而国家对其制定了相应的标准。本实验用SN/T 1391-2004 《进出口速溶咖啡检验规程》中速溶咖啡中咖啡因的测定方法测定锁阳咖啡中咖啡因的含量。http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454526_2764104_3.jpg1.方法提要样品用水溶解过滤后,用配有紫外检测器的高效液相色谱(HPLC)测定咖啡因,外标法定量。2.试剂和材料所有试剂除特殊注明外,均为分析纯,水为超纯水。2.1 乙睛:色谱纯;2.2 咖啡因标准品:纯度≥99%;2.3 标准储备液(100m g/L),准确称取 。0.0100 g 咖啡因标准品于100m L容量瓶中,用水溶解并定容至刻度,作为标准储备液。根据需要再用水将标准储备液稀释成适当浓度的标准工作液。3.仪器和设备3.1高效液相色谱仪配紫外检测器。3.2超声波振荡器。4.测定步骤4.1 提取称 取 0.1g(准确至 0.0001 g )均匀试样于 100m L容量瓶中,加人 80m L水,置超声波振荡器中超声20 min,冷却后用水定容至刻度并混匀,过0.45滤膜后,供HPLC测定。4.2 测定4 .2 .1 色谱条件a) 液 相色谱仪,配紫外检测器,检测波长273n m;b) 色 谱柱:C,。柱(25cm×4.6m mID,5um)柱或相当柱;c) 流 动相:乙睛一水一乙酸(16+83十1);d) 流 速 0.5 m L/min;e) 进 样 量 5uL4.2.2 色谱测定根据样液中咖啡因的含量情况,选定峰面积相近的标准工作溶液。标准工作溶液和样液中的咖啡因的响应值均应在仪器的检测线性范围内对标准工作溶液和样液等体积参插进样测定。在上述色谱条件下,咖啡因的保留时间约为9min。 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454528_2764104_3.jpg 标准溶液色谱图 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454529_2764104_3.jpg 标准曲线 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454530_2764104_3.jpg 样品色谱图 4.2.3[/size
影响卡尔-费休(Karl-Fisher)滴定法测水精度的几个因素方 建 安(中科院南京土壤研究所 南京 210008)一.引 言 1935年卡尔-费休(Karl-Fisher)首先提出了一种利用容量分析测定水的方法,即通常的卡尔-费休法, 它是利用碘氧化二氧化硫时需要定量的水的原理测定液体、固体和气体样法中的含水量。被许多国家定为标准分析方法, 用来校正其它分析方法和测量仪器。 因此用Karl-Fisher法测定水份含量对控制生产过程和产品质量有很好的效果。我们研制和生产的《FJA-1型常规分析仪器工作站》中的微机控制的卡尔-费休自动滴定仪的软件是根据国家标准GB/T13753-92编制而成的,它具有测定精度好,软件功能多,显示、打印和储存测定结果与曲线, 分析者可修改各种参数等特点。但测定的结果正确与否是由多种因素决定的, 除了有一个好的测定仪器外,同时考虑其它各方面的因素,才能得到可靠、正确的数据。现综合有关文献和作者的经验,把有关问题叙述如下,供有关分析者参考。二.有关问题 (一).应用范围 Karl-Fisher滴定法可适用于多种有机和无机物中含水的测定。由于各种化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。可以直接测定的主要有机和无机化合物如表1所示。 表1 无干扰的有机和无机化合物 化合物种类 举 例 1.无机化合物(1).有机酸 Na(CH3 )SO4 ,Ba(OOCCH3 )2 ,K2 C2 O4 ,VO2 (OOCCH3 )2 ,Na2 C2 H4 O6 (2).无机酸 NH4 PO4 ,CaCl2 ,NaHSO4 ,Na2 SO4 ,KF,NH4 NO3 ,MgSO4 , Na2 SO4 ,KSCN,FeSO4 ,Al2 (SO4 )3 KSO4 ,CaHPO4 , NaI,CaCO3 ,FeF3 ,VO2 (NO3 )2 (3).酸式氧化物 SiO2 ,Al2 O3 (4).无机酸和酸酐 SO2 ,HI,HF,HNO3 ,HCN,H2 SO4 ,HSO3 ,NH2 2.有机化合物(1).酸 羧酸,羧基酸,氨基酸,磺酸(2).醇 一元醇,多元醇,酚(3).酯 酸酯,正酸酯,氨基甲酸酯内酯,无机酸酯(4).稳定的羟基化合物 糖,甲醛,二苯基乙二酮,二苯乙醇酮,二氯乙醛(5).缩醛,醚 缩甲醛,二乙醚(6).烃 饱和与不饱合脂族和芳香族化合物(7).酸酐和酰卤 乙酸酐,苯甲酰氯(8).卤化物 卤代烷(9).过氧化合物 过氧化氢,二烷基过氧化物(10).含氮化合物 胺, 胺,腈(11) .含硫化合物 硫化物,硫氰酸盐,硫醚,磺原酸盐,二硫化氨基甲酸脂不能直接测定的主要有机和无机化合物如表2所示。 表2 有干扰的有机和无机化合物 化合物种类 干 扰 性 质 1.无机化合物(1).金属氢氧化物及氧化物 与费休试剂定量反应(2).碳酸盐及酸式碳酸盐 同上(3).醋酸铅,碱式氨 反应不完全(4).硼酸及氧化物 与碘反应(5).铬酸及重铬酸 非定量反应(6).钴氨络合物 同上(7).铜的氯化物及硫酸盐 被HI定量还原(8).氯化铁 与费休试剂定量反应(9).硫化氢及硫化钠 反应不确定(10).羟胺 与费休试剂部分反应(11).磷钼酸 反应不完全(12).甲基硅烷醇(R3 SiOH) 与费休试剂定量反应(13).硫代硫酸盐 同上(14).二氯化锡 同上(15).二氯化氧锆 反应不完全 2.有机化合物(1).活泼羰基化合物 形成缩醛(2).过氧化合物 与试剂中的SO2 反应(3).抗坏血酸 被碘定量氧化(4).硫醇 同上(5).醌 被HI定量还原(6).二酰基过氧化物 被HI还原(7).Dimethylo Lnred 凝聚 从上述表格中可以得出以下几点意见:1. 卡尔费休测水法适用于许多无机化合物和有机化合物中含水量的测定。 2.由于化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。因此要求分析工作者在测定某种化合物中的水时, 首先考虑它属于那一类,如果是后者,而又采用直接测定,则将产生很大的测定误差或根本无法进行测定。 3.如果要对不能进行直接测定的化合物中的水进行测定时, 必须采用合适的方法消除各种干扰因素,达到正确测定的目的。(二).仪器的标定物质卡尔费休滴定仪通常用甲醇-水标准溶液,含水酒石钠, 蒸馏水,含饱和水甲苯等类物质作为标准对方法的可靠性进行校验。含水酒石酸钠是一种常用的含水标准物质,理论含水量为15.66%,在105℃加热失重为15.65±0.02%,长期暴露于湿度为20~70%的空气中,增重为0.01~0.09%。 用含饱和水的甲苯和纯水的标定结果也是满意的。当然,最简单还是用甲醇-水标准溶液。(三).取样与取样量 在做分析取样时应尽量取混合均匀后的代表性样品,并应观察容器底部游离水分存在的情况。在用注射器抽取试样时,抽取速度不能太快,否则有可能空气进入注射器形成气泡, 造成进样误差。在分析前如果发现试样与容器有乳浊现象,或瓶壁有微小水珠析出时,则必须用乙二醇抽提法进行分析。具体方法如下: 将预先干燥的细口瓶中加入三分之一试样加盖密闭, 在工业分析天平上称准至0.1克,然后称入2至3倍于重量的乙二醇用力摇动15分种,静止分层后,用注射器通过试样层吸取0,25~1.0mL乙二醇, 测定其含水量,同时也测定乙二醇的原始水含量。分析完毕后将瓶中试样倒掉,洗净烘干,在天平上称准至0.1克,根据上述三次称量之差,求出试样和乙二醇的重量,就可求出试样的含水量。 在进样前首先用侍分析试液清洗注射器5~7次,然后根据试样含水量的多少决定取样量大小,通常按表3规定的注射器取样量抽取<0.1~5mL试样。 表3 取样量参考数据 试样含水量(ppm) 取样量(mL) 0-10 2-5 10-100 1-2 100-1000 0.1-1 >1000 <0.1 从表3中可以看到含水量大的物质取样量小,反之取样量要大,否则将产生较大的测量误差。同时特别要注意进样时注射器中是否存在小气泡, 以防产生严重的测量误差。(四).测定精度 卡尔费休滴定法测定物质含水量范围很宽从几个ppm到100%, 对精度的要求是根据含水量大小决定的。通常要求平行测定两个结果与算术平均的差数不应大于下列数值: 含水量(ppm) 允许差值 1-10 1ppm 10-50 算术平均值±10% >50 算术平均值±5% 在进行分析时,取两次测定结果的算术平均值作为分析结果。(五).影响测定精度的几个原因 除了上述测定样品的性质,测定的方法,标定物质的选用,取样方法和进样量的大小影响测定精度外,还必须注意以下几个问题,才能保证测定精度。1. 由于卡尔费休滴定试剂很容易吸收水分,因此要求滴定剂发送系统的滴定管和滴定池(测量池)等采取较好的密封系统。否则由于吸湿现象造成终点长时间的不稳定和严重的误差。2. 卡尔费休试剂的滴定度的大小, 根据试液含水量的多少来决定。在测定含水量较大的试液时,卡尔费休试剂的滴定度应该选得大一些,这样在保证测定精度(<5%)的前提下,可以加快测定速度。但在测定试液含水量较小时,卡尔费休试剂的滴定度就应该选得小一些和滴定管的最小读数小一些, 否则将产生较大的测定误差。如果滴定管的最小读数为0.01mL,卡尔费休试剂的滴定度为2.5mg/mL,则试剂一滴误差将产生0.025mg(25ppm)的测量误差。如果试剂的滴定度1.00mg/mL,则试剂一点误差将产生0.015mg(15ppm)的测量误差。 3.卡尔费休滴定法测定水的终点判别方有: (1).依靠人的视觉观察溶液颜色突变的目视法 (2).依靠观察电流表偏转突变至一定值并稳定一段时间如60秒作为滴定终点的永停终点法(硬件滴定) (3).以永停终点法又称为死停终点法(dead stop end-point method)为基础,微机自动控制的软件滴定三种方法。 目视终点法是指示终点最简单一种方法,可以省去滴定仪中的指示系统装置,在常量滴定中可以获得比较满意的测定结果,但在毫克当量以下物质的测定中,这种方法的灵敏度和准确度比较差, 一般都采用比较灵敏的电化学方法。第二种与第三种方法都是电化学方法,它有快速、灵敏而且准确度又比较高,易实现自动化等优点,通常可测定各类样品中几个ppm到百分之几十的水分。 4.滴定试剂的发送头的结构与位置也是滴定误差的一个非常重要的因素。通常要求发送滴定头内径和滴定头要做得很细,目的防止滴定剂的挂滴现象,保证测量精度。在滴定头插入样品溶液中时, 滴定头的液界处有可能发生化学反应而影响测定精度。 5.在滴定时搅拌要均充分且均匀。在滴定粘度较大的样品溶液时更要注意搅拌的充分和一致, 包括磁力搅拌器的速度要一致和滴定池中的液面高度大体相同,这样才能得到较好的测定精度。 6.在进样时,要防止注射器头受外界的污染而影响测定结果,如操作者呼气和擦注射器头时的污染等。同时要防止进样
影响卡尔-费休(Karl-Fisher)滴定法测水精度的几个因素一.引 言1935年卡尔-费休(Karl-Fisher)首先提出了一种利用容量分析测定水的方法,即通常的卡尔-费休法, 它是利用碘氧化二氧化硫时需要定量的水的原理测定液体、固体和气体样法中的含水量。被许多国家定为标准分析方法, 用来校正其它分析方法和测量仪器。 因此用Karl-Fisher法测定水份含量对控制生产过程和产品质量有很好的效果。我们研制和生产的《FJA-1型常规分析仪器工作站》中的微机控制的卡尔-费休自动滴定仪的软件是根据国家标准GB/T13753-92编制而成的,它具有测定精度好,软件功能多,显示、打印和储存测定结果与曲线, 分析者可修改各种参数等特点。但测定的结果正确与否是由多种因素决定的, 除了有一个好的测定仪器外,同时考虑其它各方面的因素,才能得到可靠、正确的数据。现综合有关文献和作者的经验,把有关问题叙述如下,供有关分析者参考。二.有关问题(一).应用范围Karl-Fisher滴定法可适用于多种有机和无机物中含水的测定。由于各种化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。可以直接测定的主要有机和无机化合物如表1所示。表1 无干扰的有机和无机化合物化合物种类 举 例1.无机化合物(1).有机酸 Na(CH3 )SO4 ,Ba(OOCCH3 )2 ,K2 C2 O4 ,VO2 (OOCCH3 )2 ,Na2 C2 H4 O6 (2).无机酸 NH4 PO4 ,CaCl2 ,NaHSO4 ,Na2 SO4 ,KF,NH4 NO3 ,MgSO4 , Na2 SO4 ,KSCN,FeSO4 ,Al2 (SO4 )3 KSO4 ,CaHPO4 , NaI,CaCO3 ,FeF3 ,VO2 (NO3 )2 (3).酸式氧化物 SiO2 ,Al2 O3 (4).无机酸和酸酐 SO2 ,HI,HF,HNO3 ,HCN,H2 SO4 ,HSO3 ,NH2 2.有机化合物(1).酸 羧酸,羧基酸,氨基酸,磺酸(2).醇 一元醇,多元醇,酚(3).酯 酸酯,正酸酯,氨基甲酸酯内酯,无机酸酯(4).稳定的羟基化合物 糖,甲醛,二苯基乙二酮,二苯乙醇酮,二氯乙醛(5).缩醛,醚 缩甲醛,二乙醚(6).烃 饱和与不饱合脂族和芳香族化合物(7).酸酐和酰卤 乙酸酐,苯甲酰氯(8).卤化物 卤代烷(9).过氧化合物 过氧化氢,二烷基过氧化物(10).含氮化合物 胺, 胺,腈(11) .含硫化合物 硫化物,硫氰酸盐,硫醚,磺原酸盐,二硫化氨基甲酸脂不能直接测定的主要有机和无机化合物如表2所示。表2 有干扰的有机和无机化合物化合物种类 干 扰 性 质1.无机化合物(1).金属氢氧化物及氧化物 与费休试剂定量反应(2).碳酸盐及酸式碳酸盐 同上(3).醋酸铅,碱式氨 反应不完全(4).硼酸及氧化物 与碘反应(5).铬酸及重铬酸 非定量反应(6).钴氨络合物 同上(7).铜的氯化物及硫酸盐 被HI定量还原(8).氯化铁 与费休试剂定量反应(9).硫化氢及硫化钠 反应不确定(10).羟胺 与费休试剂部分反应(11).磷钼酸 反应不完全(12).甲基硅烷醇(R3 SiOH) 与费休试剂定量反应(13).硫代硫酸盐 同上(14).二氯化锡 同上(15).二氯化氧锆 反应不完全2.有机化合物(1).活泼羰基化合物 形成缩醛(2).过氧化合物 与试剂中的SO2 反应(3).抗坏血酸 被碘定量氧化(4).硫醇 同上(5).醌 被HI定量还原(6).二酰基过氧化物 被HI还原(7).Dimethylo Lnred 凝聚从上述表格中可以得出以下几点意见:1. 卡尔费休测水法适用于许多无机化合物和有机化合物中含水量的测定。2.由于化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。因此要求分析工作者在测定某种化合物中的水时, 首先考虑它属于那一类,如果是后者,而又采用直接测定,则将产生很大的测定误差或根本无法进行测定。3.如果要对不能进行直接测定的化合物中的水进行测定时, 必须采用合适的方法消除各种干扰因素,达到正确测定的目的。(二).仪器的标定物质卡尔费休滴定仪通常用甲醇-水标准溶液,含水酒石钠, 蒸馏水,含饱和水甲苯等类物质作为标准对方法的可靠性进行校验。含水酒石酸钠是一种常用的含水标准物质,理论含水量为15.66%,在105℃加热失重为15.65±0.02%,长期暴露于湿度为20~70%的空气中,增重为0.01~0.09%。 用含饱和水的甲苯和纯水的标定结果也是满意的。当然,最简单还是用甲醇-水标准溶液。(三).取样与取样量在做分析取样时应尽量取混合均匀后的代表性样品,并应观察容器底部游离水分存在的情况。在用注射器抽取试样时,抽取速度不能太快,否则有可能空气进入注射器形成气泡, 造成进样误差。在分析前如果发现试样与容器有乳浊现象,或瓶壁有微小水珠析出时,则必须用乙二醇抽提法进行分析。具体方法如下:将预先干燥的细口瓶中加入三分之一试样加盖密闭, 在工业分析天平上称准至0.1克,然后称入2至3倍于重量的乙二醇用力摇动15分种,静止分层后,用注射器通过试样层吸取0,25~1.0mL乙二醇, 测定其含水量,同时也测定乙二醇的原始水含量。分析完毕后将瓶中试样倒掉,洗净烘干,在天平上称准至0.1克,根据上述三次称量之差,求出试样和乙二醇的重量,就可求出试样的含水量。在进样前首先用侍分析试液清洗注射器5~7次,然后根据试样含水量的多少决定取样量大小,通常按表3规定的注射器取样量抽取<0.1~5mL试样。表3 取样量参考数据试样含水量(ppm) 取样量(mL)0-10 2-510-100 1-2100-1000 0.1-1>1000 <0.1 从表3中可以看到含水量大的物质取样量小,反之取样量要大,否则将产生较大的测量误差。同时特别要注意进样时注射器中是否存在小气泡, 以防产生严重的测量误差。(四).测定精度卡尔费休滴定法测定物质含水量范围很宽从几个ppm到100%, 对精度的要求是根据含水量大小决定的。通常要求平行测定两个结果与算术平均的差数不应大于下列数值:含水量(ppm) 允许差值1-10 ?1ppm10-50 算术平均值±10%>50 算术平均值±5% 在进行分析时,取两次测定结果的算术平均值作为分析结果。(五).影响测定精度的几个原因除了上述测定样品的性质,测定的方法,标定物质的选用,取样方法和进样量的大小影响测定精度外,还必须注意以下几个问题,才能保证测定精度。1. 由于卡尔费休滴定试剂很容易吸收水分,因此要求滴定剂发送系统的滴定管和滴定池(测量池)等采取较好的密封系统。否则由于吸湿现象造成终点长时间的不稳定和严重的误差。2. 卡尔费休试剂的滴定度的大小, 根据试液含水量的多少来决定。在测定含水量较大的试液时,卡尔费休试剂的滴定度应该选得大一些,这样在保证测定精度(<5%)的前提下,可以加快测定速度。但在测定试液含水量较小时,卡尔费休试剂的滴定度就应该选得小一些和滴定管的最小读数小一些, 否则将产生较大的测定误差。如果滴定管的最小读数为0.01mL,卡尔费休试剂的滴定度为2.5mg/mL,则试剂一滴误差将产生0.025mg(25ppm)的测量误差。如果试剂的滴定度1.00mg/mL,则试剂一点误差将产生0.015mg(15ppm)的测量误差。3.卡尔费休滴定法测定水的终点判别方有: (1).依靠人的视觉观察溶液颜色突变的目视法 (2).依靠观察电流表偏转突变至一定值并稳定一段时间如60秒作为滴定终点的永停终点法(硬件滴定) (3).以永停终点法又称为死停终点法(dead stop end-point method)为基础,微机自动控制的软件滴定三种方法。目视终点法是指示终点最简单一种方法,可以省去滴定仪中的指示系统装置,在常量滴定中可以获得比较满意的测定结果,但在毫克当量以下物质的测定中,这种方法的灵敏度和准确度比较差, 一般都采用比较灵敏的电化学方法。第二种与第三种方法都是电化学方法,它有快速、灵敏而且准确度又比较高,易实现自动化等优点,通常可测定各类样品中几个ppm到百分之几十的水分。4.滴定试剂的发送头的结构与位置也是滴定误差的一个非常重要的因素。通常要求发送滴定头内径和滴定头要做得很细,目的防止滴定剂的挂滴现象,保证测量精度。在滴定头插入样品溶液中时, 滴定头的液界处有可能发生化学反应而影响测定精度。5.在滴定时搅拌要均充分且均匀。在滴定粘度较大的样品溶液时更要注意搅拌的充分和一致, 包括磁力搅拌器的速度要一致和滴定池中的液面高度大体相同,这样才能得到较好的测定精度。6.在进样时,要防止注射器头受外界的污染而影响测定结果,如操作者呼气和擦注射器头时的污染等。同时要防止进样
含水的纯系物为什么用色谱分离







 我要推广仪器
我要推广仪器
 下载APP
下载APP