化妆品中6-甲基香豆素的检测方法
[size=3][b]有没谁有4-甲基-5,7-二羟基香豆素的1H谱图[/b][/size]我们学校核磁仪器坏了,答辩前都修不好,希望有此化合物的图的可以给我一张,最好是扫描的。。。。谢谢了。
各位大侠帮帮忙,从文献上看到如题衍生化试剂,但上面说取适量4-溴甲基-7-甲氧基香豆素和18-冠醚-6溶于N,N-二甲基甲酰胺10ml,可到底取多少量呢,各位有知道的吗,帮帮忙,具体这个衍生化试剂怎么配得呀?急候大家的回复!谢谢啦!
苯环上3位和5位上各带一个三氟甲基,扫碳谱后非常的杂,如何才能比较准确地找出相对应的峰并计算耦合常数呢?三氟甲基上的碳是否裂分位一个4重峰了,临位的碳也被裂分为双峰了呢?
各位仁兄:三氟甲基磺酸含量如何测定?
含有三氟甲基的化合物其碳谱裂分有什么规律吗?
一看:取一块豆腐在散射光线下直接观察。传统豆腐呈均匀的乳白色或淡黄色,稍有光泽。而人工合成豆腐因为加入了白色素,比起传统豆腐要白得多,而且比较无光泽,也可能内有水纹、气泡、细微颗粒等。 二摸:传统豆腐用手按压可以感受到一定的弹性,而且软硬适度。人工合成的豆腐切面处会比较粗糙,质地不细腻,弹性较差,且没有白色豆腐液体流出。 三闻:在常温下直接嗅闻其气味。传统豆腐是以大豆为原料制成,有大豆的香味。而人工合成豆腐是用大豆分离蛋白制成,气味很淡,甚至有化学药剂的味道。

气相色谱法对维生素E的原料三甲基氢醌的检测摘要 三甲基氢醌即2,3,5-三甲基氢醌,又名2,3,5-三甲基对苯二酚,是生产维生素E的中间体,其主要用途是用作生产维生素E的主要原料。目前,维生素E已成为国际市场上用途广泛、产销量极大的主要维生素品种,国内外市场前景广阔。目前全国生产能力不能满足国内市场供应不足,部分依赖进口。因此对三甲基氢醌的需求日益增加。而对于三甲基氢醌检测目前国家和行业都没有一个统一的检测标准。为此南京科捷分析仪器应用研究所根据客户的要求应用GC5890C气相色谱仪对2,3,5-三甲基氢醌进行方法研究。实验结果表明:本方法简便,分析速度快。能满足生产质量控制的要求,从而降价低生产成本。关键词 2.3.5- 三甲基氢醌 2,3,5-三甲基对苯二酚 维生素E中间体 气相色谱法一.2.3.5三甲基氢醌气相色谱图 http://ng1.17img.cn/bbsfiles/images/2011/06/201106171053_300271_2242538_3.jpg三、仪器配置 检测项目2,3,5-三甲基氢醌及其杂质色谱仪器型号GC5890C型色谱仪 配有FID检测器毛细管色谱柱0.32*30*0.25专用柱色谱工作站N2000(电脑1台自备)氮氢空发生器 HGT300E 1台或高纯氮、氢气、空气钢瓶各一瓶
有的书上说,三氟甲基在碳谱中可产生1:3:3:1的四重峰,距离比较远。本人做过一次,基本上只得到两个较高峰,两边的小峰有扫不出来的可能吗?而且,三氟甲基对alfa碳有裂分吗?
哪位老师有检测过 2-氟-3-氯-5-三氟甲基吡啶(含量99%左右),用的什么条件和色谱柱,非常感谢!
PONY谱尼测试最新了解到,厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。PONY谱尼测试在农药残留方面具有丰富的经验,可以依据日本肯定列表进行检测。对于这三种农药,企业应该引起重视,PONY谱尼测试将鼎力帮助企业进行检测,顺利出口。
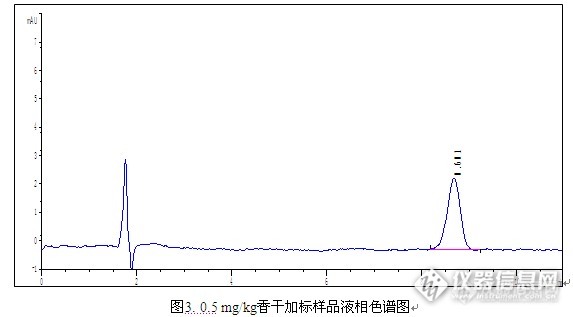
简介: 二甲基黄属于亲脂性偶氮染料,在工业上常用于油漆、鞋油、纺织品等的染色,禁止作为食品添加剂的使用。但是,近年来我国和世界各国不断报道在食品中检出工业色素二甲基黄。 二甲基黄也称甲基黄,化学名称为对二甲氨基偶氮苯,遇高温、明火或与氧化及接触有燃烧的危险。受热分解放出有毒烟气。经呼吸道、消化道、皮肤进入体内,可导致铁血红蛋白症、紫绀,严重时可致死。致癌性甲基黄是可疑人类致癌物,国际癌症研究中心已将其列入动物致癌剂。 因此通过有效的实验技术手段,对食品中的有关色素残留进行监控,以确保消费者对食品消费的安全具有重要意义。前处理方法:样品提取 称取已均质好的样品1.0 g(精确到0.01 g)试料,置于50 mL离心管中,加入15 mL甲醇,混匀后超声提取10 min,4000 r/min离心10 min,将上样液转移至另一50 mL离心管中,残渣用5 mL甲醇再次提取,离心后合并上清液,作为待净化液。样品净化 取Cleanert Sudan专用柱 (500 mg/6 mL),依次用5 mL二氯甲烷,10 mL甲醇活化小柱;将上述待净化液以1.0 ml/min流速通过小柱,再用5 mL甲醇淋洗小柱,抽干;最后用10 mL二氯甲烷洗脱,收集;40℃氮气吹干洗脱液,用1 mL甲醇溶解定容,过0.45 μm针式过滤器尼龙滤膜后,进行HPLC检测。检测条件 色谱柱:Venusil XBP C18(L),5 μm,150 Å,4.6 × 150 mm; 流动相:0.1%甲酸水溶液:甲醇= 25:75; 柱 温:30℃;波长:390 nm;进样量:20 μL;实验结果 http://ng1.17img.cn/bbsfiles/images/2015/08/201508210918_561816_2960317_3.png实验谱图 http://ng1.17img.cn/bbsfiles/images/2015/08/201508210919_561817_2960317_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508210920_561818_2960317_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508210921_561819_2960317_3.png实验结论 本实验建立了豆干中二甲基黄的检测方法,使用Cleaner Sudan专用柱对豆干样品进行前处理,当加标量为0.5 mg/kg时,平均回收率为87.4%,RSD为3.36%,说明本方法可以用于豆干中二甲基黄的检测。
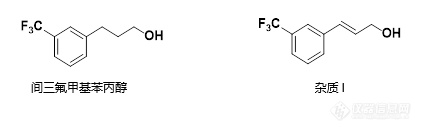
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。
三伏天喝一碗绿豆沙,健康清凉又解暑。绿豆汤中含有钾元素和维生素B1,能及时补充所流失的水分和微量元素,有效预防中暑。您知道吗?绿豆除了能做成绿豆沙、绿豆粥、绿豆饼等饮料外,还可以用来酿酒,绿豆酒口味清甜,香气浓郁,营养丰富,是夏季理想的酒精饮料。[img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308041750258963_2243_1642069_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308041750259354_4925_1642069_3.png[/img]
跟着5009.284-2021走的梯度洗脱,4个标准品配的混标,只有甲基香兰素和乙基香兰素分不开,呈大M,自己试了好几种梯度都分不开,色谱柱是全新的,排除色谱柱原因,想问一下大家有没有更好的梯度洗脱?[img]https://ng1.17img.cn/bbsfiles/images/2022/08/202208311552565427_9283_5511445_3.jpeg[/img]
我想了解4-氯-2-三氟甲基苯腈(4-Chloro-2-(trifluoromethyl)benzonitrile,CAS#320-41-2)的理化性质,但在网上只找到沸点109 º C (10 mmHg),是液体还是固体看不出来。因为这个沸点是真空条件下的。那位老师有相关的信息,请告诉我,谢谢!
求助二氟甲基-2,2,2-三氟乙基醚的标准红外谱图,谢谢上传。
我家喜欢各种各样的浇汁菜,例如浇汁鱼、浇汁豆腐、浇汁蒸蛋、浇汁青菜等等,所谓浇汁菜,我自己的理解是,将各种配菜或配料炒制或焖制后浇在提前处理好的主料上,这样做出来的菜味道格外的香浓和入味,而且选择性也多,今天给大家推荐的是一道三鲜浇汁豆腐,选用了比一般豆腐口感更嫩一些的日本豆腐,裹上蛋涂后提前炸好,浇上汁的那一刹那那是绝对的香味四溢,而且这个菜特别受小朋友欢迎,因为除了豆腐,虾仁、胡萝卜和香菇都是他们喜欢的食材,而且口味香浓中又不乏清淡。 材料:日本豆腐4个、胡萝卜半根、香菇几朵、鲜虾100克、鸡蛋1个、鲜红椒1个、姜、香葱、植物油、盐、高汤、水淀粉 做法:1、新鲜的虾剥去虾壳,挑去虾线,洗干净后切成小段 2、香菇、胡萝卜分别切丁、姜切丝、辣椒切米、葱切花 3、取出日本豆腐,一个切成五段,取一个鸡蛋打散,蛋液里加少许盐调味 4、日本豆腐放入蛋液中,裹上薄薄的一层,入锅炸至金黄色 5、炸好后捞出来沥干净油后放入盘中备用 6、重新起锅放油烧热,放入姜丝爆香,然后放入胡萝卜丁翻炒一会儿 7、再放入虾仁、香菇和红椒一同翻炒 8、炒至虾仁变色后加适量盐调味 9、加入小半碗高汤,小焖片刻 10、加入适量水淀粉勾个薄欠,待汤汁变浓变少时,起锅浇在事先炸好的豆腐上即可。
我想问一下三氟甲基亚磺酸钠的液谱分析方法,哪位高手请指点一下!谢谢

[align=center][img=,600,400]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091122509261_9069_932_3.jpg!w600x400.jpg[/img][/align]香兰素、甲基香兰素和乙基香兰素,均为广泛使用的可食用香料。有浓烈的奶香气息,在香荚兰的种子中可以找到,也可以人工合成,被广泛的运用到,蛋糕、奶粉、冰激凌等食品的制作中。今天我们就来做一下在奶粉和蛋白粉中香兰素、甲基香兰素和乙基香兰素的检测。[b]适用范围[/b]适用于奶粉、蛋白粉中香兰素、甲基香兰素和乙基香兰素的检测。[b]溶液的配置[/b]1)标准储备液:分别精确称取香兰素、甲基香兰素和乙基香兰素10mg,分别用乙腈溶解并定容到10mL,浓度为1000mg/L。2)20%甲醇:移取20mL的甲醇,用水定容至100mL。3)5%乙酸甲醇:移取5mL乙酸,用甲醇定容至100mL。4)50%乙腈:移取50mL的乙腈,用水定容至100mL。[b]提取步骤[/b]奶粉1) 移取1g样品,加入10mL的水,振荡,超声10min,离心10min(8000r/min),吸取上清液;2) 再加入10mL水,振荡,超声10min,离心10min(8000r/min),吸取上清液;3) 重复2)的过程,合并所有上清液,混匀,再次离心2min,待净化。蛋白粉移取0.5g样品,加入10mL的水振荡,8000rpm下离心,取澄清液,再重复2次,总计30mL水提取,待净化。[b]SPE净化步骤[/b]SPE柱:月旭WelchromP-SAX规格:150 mg/6mL。活化:5 mL 甲醇、5 mL 水,弃去;上样:待净化液15mL上样,控制流速,不宜过快,弃去;淋洗:6mL20%甲醇水淋洗,弃去。洗脱:15mL5%乙酸甲醇洗脱,收集于旋转蒸发瓶,并抽干小柱。复溶:洗脱液在45℃下减压蒸干,用50%乙腈定容至1mL。过0.22μm滤膜,上HPLC检测。[b]色谱条件[/b][color=#333333][/color]色谱柱:月旭UltimateXB-C18 4.6×250mm,5μm流动相:A-0.1%磷酸溶液,B-甲醇(A/B=70/30等度洗脱)流速:1.0mL/min柱温:30℃进样量:20μL检测波长:280nm[align=left][b]色谱图或者加标回收率结果[/b][/align][align=center][b][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091122549495_1545_932_3.jpg!w690x386.jpg[/img][/b][/align][align=center][color=#333333][img=,600,131]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091122584398_3527_932_3.png!w690x151.jpg[/img][/color][/align][align=center][color=#333333]图1.奶粉对照香兰素20mg/L、甲基香兰素5mg/L、乙基香兰素10mg/L图谱[/color][/align][color=#333333][/color][align=center][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123014375_3369_932_3.jpg!w690x386.jpg[/img][/color][/align][align=center][color=#333333]图2.奶粉粉样过柱图谱[/color][/align][align=center][color=#333333][/color][/align][align=center][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123043784_5835_932_3.jpg!w690x386.jpg[/img][/color][/align][align=center][color=#333333]图3.奶粉样加标香兰素20mg/L、甲基香兰素5mg/L、乙基香兰素10mg/L图谱[/color][/align][align=center][color=#333333][/color][/align][align=center][color=#333333][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123079138_6486_932_3.jpg!w690x386.jpg[/img][/color][/color][/align][align=center][color=#333333][color=#333333][img=,600,158]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123107145_4550_932_3.png!w690x182.jpg[/img][/color][/color][/align][align=center][color=#333333][color=#333333]图4.蛋白粉对照香兰素100mg/L、甲基香兰素5mg/L、乙基香兰素15mg/L图谱[/color][/color][/align][align=center][color=#333333][color=#333333][/color][/color][/align][align=center][color=#333333][color=#333333][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123138565_2990_932_3.jpg!w690x386.jpg[/img][/color][/color][/color][/align][align=center][color=#333333][color=#333333][color=#333333]图5.蛋白粉样过柱图谱[/color][/color][/color][/align][color=#333333][color=#333333][color=#333333][/color][/color][/color][align=center][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123171107_510_932_3.jpg!w690x386.jpg[/img][/align][align=center]图6.蛋白粉样加标香兰素400mg/kg、甲基香兰素20mg/kg、乙基香兰素60mg/kg图谱[/align][align=center][/align][align=center][img=,600,216]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123204485_8036_932_3.png!w561x202.jpg[/img][/align][color=#333333][/color][align=center]表1.奶粉香兰素、甲基香兰素和乙基香兰素过P-SAX小柱加标回收表[/align][align=center][/align][align=center][color=#333333][color=#333333][color=#333333][img=,600,162]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123255715_3868_932_3.png!w641x174.jpg[/img][/color][/color][/color][/align][align=center][color=#333333]表2.蛋白粉香兰素、甲基香兰素和乙基香兰素过P-SAX小柱加标回收表[/color][/align][color=#333333][color=#333333][color=#333333][b]相关产品信息[/b][/color][/color][/color][align=center][img=,600,407]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123287959_7092_932_3.jpg!w690x469.jpg[/img][/align]
5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。
豆油皮,素肉,腐竹这三个做蛋白质,蛋白换算系数一样吗?

“超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留”是本人去年开展大豆中草甘膦检测项目整个试验过程的总结,欢迎各位老师和同行批评指正,该文章还未在任何刊物上发表。[align=center][b]超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留[/b][/align][align=center][/align][align=center]户江涛[/align][align=center](黑龙江省农垦科学院测试化验中心,黑龙江 佳木斯 154007 )[/align]摘要:采用超高效液相色谱-串联质谱法建立了快速检测大豆中草甘膦和氨甲基膦酸残留量的分析方法。试样经水超声提取,二氯甲烷去除脂肪,C[sub]18[/sub]固相萃取柱净化后,在硼酸钠缓冲溶液中与9-芴甲氧羰酰氯(FMOC-Cl)进行衍生反应,其衍生产物在C[sub]18[/sub]色谱柱上以 2 mmol/L 乙酸铵溶液和乙腈为流动相,进行液相色谱分离:质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明,草甘膦和氨甲基膦酸在0.001~0.5 mg/L范围内线性关系良好,相关系数(R)分别为0.9996和0.9993,定量限(LOQ)均为0.01mg/kg。在空白大豆样品添加浓度为0.02、0.2、2 mg/kg 时,草甘膦和氨甲基膦酸的平均回收率分别为80.2%~91.5%和77.7%~89.3%,相对标准偏差(RSD)分别为3.37%~6.96%和4.11%~8.27%(n=6)。本方法快速、简便、灵敏,适用于大豆中草甘膦和氨甲基膦酸残留的同时检测。关键词:超高效液相色谱-串联质谱;大豆;草甘膦;氨甲基膦酸;衍生反应[align=center]Determination of glyphosate and its metabolite aminomethyl-phosphonic acid residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry[/align][align=center]HU Jiangtao[/align][align=center]([i]Testing and Analysis Center of Heilongjiang Academy of Land Reclamation Sciences, Jiamusi 154007,China[/i])[/align][b]Abstract:[/b]A method[b] [/b]was developed for the determination of glyphosate(PMG) and aminomethyl-phosphonic acid(APMA) residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). After extracted with water under ultrasonication, the sample was defatted with dichloromethane and purified by C[sub]18 [/sub]solid phase extraction cartridge, and then PMG and APMA were derivatized using 9-fluorenylmethoxycarbonyl(FMOC-Cl) in borate buffer for 2 h.The derivatives of PMG and APMA were separated on a Waters BEH C[sub]18[/sub] column with gradient elution with the mobile phase of 2 mmol/L ammonium acetate and acetonitrile, and finally detected by positive eletrospray ionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reaction monitoring(MRM) mode.The results showed the linearities of PMG and APMA were good in the concentration range of 0.001~0.5 mg/L ,and the correlation coefficients were 0.9996 and 0.9993 respectively. The limit of quantification(LOQ) of PMG and APMA was both 0.01mg/kg. At the spiked levels of 0.02、0.2、2 mg/kg in the blank soybean samples, the mean recoveries of PMG and APMA were 80.2%~91.5% and 77.7%~89.3% respectively, and the relative standard deviation(RSD) of PMG and APMA were 3.37%~6.96% and 4.11%~8.27% res-pectively(n=6).This method is fast,simple,sensitive, and suitable for simultaneous determination of PMG and APMA in soybean.[b]Key words: [/b]ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) soybean glyphosate(PMG) aminomethyl-phosphonic acid(APMA) derivatization草甘膦(Glyphosate,PMG)又名镇草宁、农达,分子式为C[sub]3[/sub]H[sub]8[/sub]NO[sub]5[/sub]P,是1971年美国孟山都公司研发的一种有机磷除草剂,因其兼具内吸、传导性、灭生性及非选择性,同时不易在生物体内累积,故广泛应用于农业生产中一年生和多年生杂草防除,是目前世界上应用最广、生产量最大的除草剂[sup][/sup]。草甘膦及其在植物中的主要代谢物氨甲基膦酸(Aminomethyl-phosphonic acid,APMA,分子式为CH[sub]6[/sub]NO[sub]3[/sub]P)均属于强极性、易溶于水的高沸点化合物,具有不易挥发、无紫外吸收等特性,因此用常规方法分析检测十分困难[sup][/sup]。 目前, PMG和APMA残留检测的方法主要有色谱法(GC[sup][/sup]、LC[sup][/sup]、IC[sup][/sup])、质谱法(GC/MS[sup][/sup]、ICP/MS[sup][/sup]、LC/MS/MS[sup][/sup])、光谱法[sup] [/sup]等。光谱法虽然操作简便,但其灵敏度不高,而[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法[sup][/sup]只能适用于水样等简单基质,用于植物源样品检测时干扰太大;用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]技术检测时,需要将PMG和APMA衍生转化为可气化物质,其引入试剂多、过程相对繁琐,效率较低[sup][/sup];用LC/MS/MS法直接检测时[sup][/sup],由于PMG和APMA分子量(分别为169、111)均较小,其主要碎片离子的质荷比多在100以下,检测实际样品时受基质干扰严重,灵敏度较低,因此柱前衍生——[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]法成为近年来国内外检测PMG和APMA残留的主流方法[sup][/sup]。以9-芴甲氧羰酰氯(FMOC-Cl)做为衍生试剂,在硼酸盐缓冲溶液中与PMG和APMA水提取液相容性好,过程简单,其衍生产物在LC/MS/MS中响应信号高,碎片离子干扰小,适合定性定量分析。 目前,采用柱前衍生——LC/MS/MS法检测茶叶、稻米等基质中PMG和APMA残留的报道很多[sup][/sup],专门针对大豆基质的报道很少。行业标准[sup][/sup]的适用范围虽然包括了大豆基质,但该方法在实验过程中试剂用量大、操作繁琐(反复调pH值)、衍生时间长(需过夜),尤其是使用阳离子交换柱(CAX)洗脱时需要加入11 mL 1%的盐酸甲醇水(20/80,v/v),水分含量过高导致旋转蒸发时很难蒸干,容易造成PMG和APMA回收率不稳定。本文专门针对大豆这类高蛋白、高脂肪含量的特殊基质,采用纯水作为提取试剂,二氯甲烷去除脂溶性杂质,C[sub]18[/sub]固相萃取小柱净化后采用FMOC-Cl衍生,最后用UPLC/MS/MS测定。该方法前处理过程简便、快速、灵敏度高,适用于大豆中PMG和APMA的残留检测。[b]1 实验部分[/b]1.1 材料与试剂 草甘膦、氨甲基膦酸(纯度≥99%,德国Dr.Ehrensorfer公司);FMOC-Cl(纯度99%,Sigma公司),使用时配置成10g/L的丙酮溶液;乙腈、二氯甲烷、甲酸、乙酸铵(色谱纯,美国Fisher公司);十水四硼酸钠(优级纯,天津市科密欧化学试剂有限公司),使用时配置成50g/L的水溶液;实验用水为Millipore纯水仪制备;C[sub]18[/sub]固相萃取小柱(200mg/3ml,美国Agilent公司)。1.2 仪器与设备 Acquity UPLC型超高效液相色谱仪(Waters公司);XEVO TQ-S三重四级杆质谱仪(Waters公司);CR21GⅢ型高速离心机(HITACHI公司);KQ5200DB型台式超声波仪(昆山市超声仪器有限公司);涡旋混合器(IKA公司)。1.3 标准溶液的配置 分别称取草甘膦和氨甲基膦酸标准品10mg(精确到0.1mg),用水溶解并定容至10mL,配置成质量浓度为1.0 mg/mL标准储备液,于4℃冰箱保存待用;使用时用水逐级稀释成所需浓度的混合标准工作液。1.4 样品前处理 提取:称取粉碎均匀后的试样1.0g(精确到0.01g)于50mL聚乙烯离心管中,加入10.0mL超纯水,涡旋混合30 s并超声提取20 min后,以10000 r/min离心3 min,将上清液转移至另一离心管中,加入5 mL二氯甲烷涡旋混合30 s,以10000 r/min离心3 min,上清液待净化。 净化:取2.5 mL上清液加入到C[sub]18[/sub]固相萃取柱(使用前依次用3mL甲醇和3mL超纯水活化)中,弃去最初的几滴流出液(约0.5 mL),将剩余部分用5 mL玻璃管收集,待衍生。 衍生:取1.0 mL净化液于5 mL离心管中,依次加入1.0 mL 50g/L的硼酸钠溶液和 1.0 mL 10g/L的FMOC-Cl衍生液,混匀后室温下衍生2 h,以10000 r/min离心3 min,取上清液过0.22 mm有机系微孔滤膜后,供UPLC/MS/MS分析测定。1.5 液相色谱及质谱条件 液相色谱:色谱柱:Waters BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm);柱温:30℃;流速:0.5 mL/min;进样量:2 μL;流动相A:乙腈;流动相B: 2 mmol /L 的乙酸铵水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1min,100% A~10% A,4.1 ~5.0min 10% A。 质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1 PMG-FMOC和 AMPA-FMOC的MRM质谱参数[/align][align=center]Table 1 MRM parameters of PMG-FMOC and AMPA-FMOC[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Cone/V[/align][/td][td][align=center]Parent ion/(m/z)[/align][/td][td][align=center]Daughter ion/(m/z) [/align][/td][td][align=center]Collision energy/V[/align][/td][/tr][tr][td][align=center]PMG-FMOC[/align][align=center] [/align][align=center]AMPA-FMOC[/align][/td][td][align=center]30[/align][align=center] [/align][align=center]30[/align][/td][td][align=center]392[/align][align=center][sup] [/sup][/align][align=center]334[/align][align=center][sup] [/sup][/align][/td][td][align=center]88[/align][align=center]214﹡[/align][align=center]112﹡[/align][align=center]179[/align][/td][td][align=center]14[/align][align=center]8[/align][align=center]11[/align][align=center]20[/align][/td][/tr][/table]﹡quantitative ion[b]2 结果与讨论[/b]2.1 色谱及质谱条件的优化 流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与非酸性体系(乙酸铵水溶液)分别于甲醇、乙腈的流动相体系组合,结果发现两种分析物在酸性体系中分离效果欠佳,峰形拖尾严重,而在非酸性体系中其色谱分离效果得到明显改善,峰形对称;乙腈比甲醇体系洗脱能力更强,可以有效缩短分析时间。故本研究采用乙酸铵水溶液+乙腈流动相体系,并比较了1、2、5 mmol/L三种乙酸铵浓度与乙腈的组合,结果发现随着乙酸铵浓度的增加,目标物响应值虽略有提高但相差不大,而同时仪器背景值却显著升高,综合考虑目标物信号强度、信噪比、色谱分离效果以及分析时间等因素,本实验最终选择了2 mmol /L 乙酸铵水溶液+乙腈分析体系。质谱的选择:PMG、 AMPA对应的衍生物PMG-FMOC、AMPA-FMOC分子量分别为391、333。用超纯水配置10 mg/L 混合标准溶液直接注射到质谱中,在正负离子模式下分别进行母离子全扫描,发现正离子模式下392、334具有很好的响应,然后分别以392、334为母离子进行子离子全扫描,各得到两组丰度高、干扰小的子离子对进行MRM监测,最终确定的质谱条件见表1。2.2 前处理条件的优化 提取溶液的选择:PMG和APMA属于强极性物质,易溶于水,难溶于有机溶剂,故一般采用极性溶剂提取,如纯水及KOH、NaHCO[sub]3[/sub]溶液等[sup][/sup]。实验发现,用碱性溶液提取后,大豆中脂肪、蛋白等物质会与碱性物质发生反应,导致离心后的提取液异常浑浊,不利于后期净化和衍生,因此本实验采用纯水作为提取试剂,再经二氯甲烷液液萃取去除脂溶性杂质。 净化柱的选择:研究发现,对提取后的溶液不经SPE净化直接进行衍生, PMG和APMA的回收率均不足30%,且精密度很差,这可能是由于大豆中富含脂肪、蛋白质等物质干扰衍生过程,故本实验比较了对脂肪、蛋白质有很好去除效果的C[sub]18[/sub]、中性Al[sub]2[/sub]O[sub]3[/sub]、HLB固相萃取SPE柱的净化效果,结果发现提取液经中性Al[sub]2[/sub]O[sub]3[/sub]净化后,PMG和APMA几乎检测不到;而C[sub]18[/sub]净化后目标物回收率为92.7%、90.8%,HLB为83.6%、80.5%。故本实验选取了净化效果更好,成本相对低廉的C[sub]18[/sub]固相萃取小柱。 衍生条件的优化:FMOC-Cl的衍生机制是在碱性环境下(pH=9.0)通过FMOC-Cl基团取代目标化合物氮原子上的氢,从而生成较稳定的化合物FMOC-Cl。参照行业标准[sup][/sup]及文献报道[sup][/sup]所选用的缓冲液浓度,本实验采用50g/L的硼酸钠水溶液缓冲液体系,设置的衍生试剂质量浓度为1、2、5、10、20 g/L FMOC-Cl丙酮溶液,按照本文1.4步骤对PMG和APMA质量浓度为0.5 mg/L的纯水溶液和大豆空白基质溶液分别进行衍生,结果见图1。结果表明,在纯水溶液中,FMOC-Cl浓度为2 g/L时,PMG和APMA的峰面积已达到最大,随着衍生化试剂浓度的升高,其峰面积无明显变化;而在大豆空白基质溶液中,FMOC-Cl低浓度(1、2g/L)时,PMG和APMA几乎检测不到,其峰面积随衍生化试剂浓度增加而加大,浓度到达一定程度(10 g/L)时,峰面积不再变化。产生这种现象的原因,可能是由于尽管大豆提取液经过了二氯甲烷和C[sub]18[/sub]小柱的净化,但还是会有少量水溶性蛋白、脂肪等杂质残留在净化液中,这些杂质可能会与衍生试剂反应,影响目标物的衍生效果。研究还发现,当FMOC-Cl浓度为20 g/L时,得到的PMG和APMA色谱峰产生拖尾现象,可能是由于衍生试剂化学性质较活泼,其用量大时,过量的FMOC-Cl会迅速转化成FMOC-OH,干扰目标物峰形。在50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液条件下,考察不同时间(0.5h、1h、2h、4h、8h和16h)对衍生效果的影响,结果发现,2 h后PMG和APMA的测定值无明显增加。因此,本实验最终选定的衍生条件为50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液,室温下衍生2 h。[img=,596,378]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_01_2984502_3.png[/img][img=,690,530]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_02_2984502_3.png[/img]2.3 基质效应的考察 基质效应(主要是抑制)是LC/MS/MS仪器检测时经常遇到的现象。由于本实验采用极性很强的水作为提取剂,大豆中的色素、脂肪酸等极性较强的物质也有少部分进入到最后的上机液中,在离子化带电过程中会与目标物产生竞争,抑制目标物的离子化效率。实验考察了用PMG和APMA的纯水标样去标定经过本文1.4步骤处理后的大豆空白基质溶液配置的同浓度标样,其色谱图见图2。结果发现,PMG在纯水和大豆空白基质中峰面积基本一致,而APMA在大豆空白基质中的峰面积仅为纯水中的55.7%,产生了明显的基质抑制效应。为了消除基质干扰,本实验选用大豆样品空白基质配置不同浓度的标准溶液来绘制标准曲线进行校准。2.4 线性范围和定量限 用大豆空白基质溶液分别配置0.001、0.005、0.01、0.05、0.1、0.2、0.5 mg/L的PMG和APMA混合标准溶液,按本文1.4步骤衍生后测定,以各自定量离子的峰面积为Y对应质量浓度X(mg/L)做标准曲线,得到的线性方程和相关系数见表2。结果表明,这两种物质在0.001~0.5 mg/L浓度范围内线性良好,相关系数R分别为0.9996和0.9993。以10倍信噪比(S/N)计算,该方法PMG和APMA的定量限(LOQ)均为0.01 mg/kg。[align=center]表2 PMG和APMA大豆基质标准溶液的线性方程、相关系数和定量限(LOQ)[/align][align=center]Table 2 Linear equations,correlation and LOQ of PMG and APMA in the soybean matrix standard solutions[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Linear range/(mg/L)[/align][/td][td][align=center]Linear equation[/align][/td][td][align=center]R[/align][/td][td][align=center]LOQ/(mg/ kg )[/align][/td][/tr][tr][td][align=center]PMG[/align][align=center]AMPA[/align][/td][td][align=center][sup]0.001~0.5[/sup][/align][align=center][sup]0.001~0.5[/sup][/align][/td][td][align=center]Y=889809x+1671.3[/align][align=center]Y=476982x+1161.9[/align][/td][td][align=center]0.9996[/align][align=center]0.9993[/align][/td][td][align=center]0.01[/align][align=center]0.01[/align][/td][/tr][/table]2.5 回收率和精密度 称取大豆空白试样1.0 g,分别添加0.02、0.2、2 mg/kg水平的PMG和APMA混合标样,每个水平重复6次,按照本文1.4步骤前处理方法处理后上机检测,实验结果见表3。从表3可以看出,PMG的平均回收率为80.2%~91.5%,相对标准偏差(RSD,n=6)为3.37%~6.96%;APMA的平均回收率和RSD分别为77.7%~89.3%和4.11%~8.27%。[align=center]表3 大豆中PMG和APMA的加标回收率和相对标准偏差(n=6)[/align][align=center]Table 3 Recoveries and relative standard deviations(RSD)of PMG and APMA spiked in the soybean(n=6) [/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Spiked level(mg/kg)[/align][/td][td][align=center]Recovery/%[/align][/td][td][align=center]RSD/%[/align][/td][/tr][tr][td]PMGAMPA[/td][td][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][/td][td][align=center]80.2[/align][align=center]91.5[/align][align=center]86.8[/align][align=center]77.7[/align][align=center]89.3[/align][align=center]85.9[/align][/td][td][align=center]6.96[/align][align=center]3.37[/align][align=center]3.95[/align][align=center]8.27[/align][align=center]4.25[/align][align=center]4.11[/align][/td][/tr][/table][b]3 结语[/b] 本文建立了超高效液相色谱-串联质谱法(UPLC/MS/MS)测定大豆中草甘膦及其代谢物氨甲基膦酸残留的分析方法。该方法灵敏度高,PMG和APMA定量限(LOQ)达到0.01 mg/kg,能满足大豆产品相关限量标准要求。同时该方法具有较高的准确度和精密度,前处理步骤简单快速,特别适合大批量大豆样品的检测。
各位老师好!想请教下香豆素和异香豆素极性的大小,在c18柱子上的出峰顺序,感谢!
二氢香豆素中的有时会含有痕量香豆素,用极性柱子测定时,二氢香豆素拖尾较严重,有时会影响香豆素的定量。哪位网友有什么好的方法(什么柱子和条件)能让二氢香豆素德拖尾减少,并与香豆素分离更好?
在用固相微萃取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测甲硫醚和二甲基三硫醚时,为什么二甲基三硫醚老是不出啊
[size=18px]目前在用AB的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测三苯基氯甲烷,Q1 MI模式扫243.1的离子[font=-apple-system, BlinkMacSystemFont, &](应该是三苯甲基碳正离子)[/font],发现基线非常高(30万-50万之间),且不稳定,时高时低,导致峰面积也 不稳定,打电话问客服,几个人几种说法,“液相部分污染了”“这个是正常现象,多走走就稳定了”,尝试用MRM模式去做,打出一个165.2的碎片,基线不到1000,做了线性和回收也都挺好,但是,这个碎片离子是怎么打出来的比较困惑,就怕以后再做的时候重现不出来……[/size][size=18px]流动相是90%甲醇,溶剂是正丁醇:乙腈(80:20)[/size][size=18px]请教一下各位大神,AB的仪器用SIM模式选择Q1 MI还是Q3 MI好呢?基线高且时高时低,除了污染还有什么原因呢?[font=-apple-system, BlinkMacSystemFont, &]三苯甲基碳正离子在质谱里能被打碎吗?会裂解成什么碎片离子?[/font][/size][size=18px][font=-apple-system, BlinkMacSystemFont, &][/font][/size]
各位老师 我想 求一下 甲基肼, 三氯化磷,磷化氢的标准曲线和相对应的吸光度 谢谢 甲基肼-对二甲胺基苯甲醛分光光度法 三氯化磷-对氨基二甲基苯胺分光光度法 磷化氢-钼酸铵分光光度法 五氧化二磷-钼酸铵分光光度法谢谢各位老师
氨基三亚甲基膦酸 ATMPAmino Trimethylene Phosphonic Acid【CAS】 6419-19-8别名:氨基三甲叉膦酸 Dequest 2000分子式 N(CH2PO3H2)3 相对分子质量:299.05一、性能与用途本品具有良好的螯合、低限抑制及晶格畸变作用。可阻止水中成垢盐类形成水垢,特别是碳酸钙垢的形成。ATMP在水中化学性质稳定,不易水解。在水中浓度较高时,有良好的缓蚀效果。本品用于火力发电厂、炼油厂的循环冷却水、油田回注水系统。可以起到减少金属设备或管路腐蚀和结垢的作用。本品在纺织印染等行业用作金属离子螯合剂,也可用于金属表面处理剂等。二、质量指标 符合HG/T 2841-2005 符合HG/T 2841-1997 符合HG/T 2841-2005项 目 指 标外 观 无色或淡黄色透明液体活性组分(以ATMP计) % ≥ 50.0 50.0氨基三亚甲基磷酸含量% ≥ —— 40.0亚磷酸(以PO33-计) % ≤ 5.0 3.5磷酸 (以PO43-计) % ≤ 1.0 0.8PH值(1%水溶液) 1.5-2.5 1.5-2.5 氯化物 (以Cl-计)% ≤ 3.5 2.0Fe(以Fe3+)含量ppm ≤ —— 20.0 密度(20℃) g/cm3 ≥ 1.28 1.30三、应用范围与使用方法常与其它有机膦酸、聚羧酸或盐等复配成有机碱性水处理剂,用于各种不同水质条件下的循环冷却水系统。用量以1~20mg/L为佳;作缓蚀剂使用时,用量为20~60mg/L。四、包装与贮存塑料桶包装,每桶30Kg 或250Kg,也可根据用户需要确定。贮于室内阴凉处,贮存期十个月。五、安全防护:本品为酸性,应避免与眼睛、皮肤或衣服接触,一旦溅到身上,应立即用大量水冲洗。







 我要推广仪器
我要推广仪器
 下载APP
下载APP