甲基环戊二烯三羰基锰(MMT)气相色谱法检测方法本标准规定了甲基环戊二烯三羰基锰的分类、要求、试验方法、检验规则、标志、包装、运输、贮 存和安全。本标准适用于用作汽油抗爆剂的甲基环戊二烯三羰基锰。 分子式:C9H7MnO3 相对分子质量:218.09(根据2007年国际相对原子质量) 甲基环戊二烯三羰基锰含量的测定:在选定的工作条件下,样品经气化通过毛细管色谱柱,使其中各组分得到分离,用氢火焰离子化检 测器检测,用面积归一化法或内标法计算甲基环戊二烯三羰基锰的含量。 试剂:二乙二醇二甲醚。 无水乙醇。氢气:体积分数不低于 99.99%。 空气:经活性炭和分子筛净化。氦气:体积分数不低于 99.999%。仪器设备 :GC5890气相色谱仪,配氢火焰离子化检测器(FID),灵敏度和稳定性符合 GB/T9722 中的有关规定, 可进行毛细管色谱分析。N2000色谱工作站。色谱仪器型号GC5890型色谱仪 配有FID检测器毛细管色谱柱HP-5 30*0.32*0.25专用毛细管柱色谱工作站N2000 (电脑1台自备)气体装置氮氢空发生器 HGT300E1台或高纯氮、氢气、空气钢瓶各一瓶分析天平:感量 0.0001g。 5.8.3.4 进样器:5μL [font=
[em0808] [em0808] 各位求助一下关于2.5-二甲基-2.5-双(叔丁基过氧基)己烷的测定,性质,反正关于2.5-二甲基-2.5-双(叔丁基过氧基)己烷的任何资料都可以了,在网上都查过了,找到一篇,我先发上来大家看看.有的话麻烦大家分享一下了[~82374~]
叔丁基甲基醚,分析纯,不知道国内是否把叔丁基甲基醚 按照色谱纯,分析纯,化学纯来分的?还是直接按照纯度来分(比如≥99%)因为问过了国药之类的试剂商,都没叔丁基甲基醚 分析纯这一说法≥99%纯度的叔丁基甲基醚 和分析纯的叔丁基甲基醚 发现差别太多了,里面的杂质之类的,包括价格,,求解释?最好有人能提供试剂商,不知道去哪买叔丁基甲基醚了。
[color=#444444]ESI 质谱条件下,M+H的二级产生含羰基的加氢峰,m/z 220,M+Na的二级碎片产生含羟基的加钠峰m/z 244,二者相差24。羰基氧和羟基氧与钠离子和氢离子的结合能力是怎样?[/color][color=#444444]难道钠离子更容易稳定含羟基离子?氢离子更容易稳定含羰基的离子?该怎么解释呢?[/color]
在外文文献中,看到用甲基叔丁醚活化萃取柱,净化农药,不知道有没有它的替代品,比如说异辛烷、环己烷等。谢谢诶
![N-[芴甲氧羰基]-N-甲基-S-(三苯基甲基)-L-半胱氨酸|Fmoc-N-Me-Cys(Trt)-OH](https://ng1.17img.cn/bbsfiles/images/2021/02/202102031611349730_9244_3531468_3.jpg)
中文名称:N-[芴甲氧羰基]-N-甲基-S-(三苯基甲基)-L-半胱氨酸英文名称:Fmoc-N-Me-Cys(Trt)-OHCAS号:944797-51-7【详情请咨询国肽生物】别名:L-Cysteine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-N-methyl-S-(triphenylmethyl)-Fmoc-MeCys(Trt)-OH Fmoc-N-Me-L-Cys(Trt)-OH 分子式:C38H33NO4S分子量:599.73800结构图:多肽合成主要是采用Fmoc合成法。Fmoc合成法采用Fmoc为α-氨基的保护基,侧链保护采用苄醇类。合成时将一个Fmoc-氨基酸衍生物共价交联到树脂上,用碱脱除Fmoc,用三乙胺中和游离的氨基末端,然后通过DCC活化、偶联下一个氨基酸,脱保护多采用HF法或TFMSA(三氟甲磺酸)法。多肽合成服务种类多肽合成服务通常有线性肽合成服务、多种难肽合成服务、修饰肽合成服务、以及部分多肽合成公司还会提供多肽定制服务,定制出有针对性的合成肽。目前有多肽合成公司提供的线性肽合成可达150个氨基酸以内,在修饰肽合成上,能提供常见修饰,磷酸肽,RGD环肽,荧光标记肽(Cy3、Cy5、Fitc、AMC等),生物素标记肽/复合抗原(MAP)/含D型氨基酸,及各种氨基酸衍生物均可合成。多肽产物纯度选择常见的质谱级多肽纯度,一般要求95%用于抗体筛选纯度,一般85%即可NMR和结晶试验中,纯度一般98%粗品肽,一般50%即可用于多肽筛选多肽多肽是一种与生物体内各种细胞功能都相关的生物活性物质,它的分子结构介于氨基酸和蛋白质之间,是由多种氨基酸按照一定的排列顺序通过肽键结合而成的化合物。多肽是涉及生物体内各种细胞功能的生物活性物质的总称,常常被应用于功能分析、抗体研究、尤其是药物研发等领域。多肽合成技术的出现,让这些多肽的应用领域变得更宽。供应高品质普通多肽我们拥有成熟的多肽合成纯化方法,利用SPPS方法和液相合成方法为客户提供高品质多肽。我们的服务特点是:1. 纯度:我们提供粗品肽和纯度纯度为70%,75%,80%,85%,90%,95%,98%,99%的纯品多肽。2.脱盐和转盐:根据客户要求,我们可以对多肽进行脱TFA盐处理,也可以转为醋酸盐。3.交货期限:30个氨基酸之内,一般2-3周,最快1-2周。4.质量控制:每条多肽都免费提供合格的HPLC,MS和COA文件。5.售后服务:1-2周内可以提出异议,我们免费复测,不合格免费退货,1-3个月内使用不合格可以免费提供复测,样品免费保存3个月。供应各种修饰型多肽1.磷酸化的Ser、Tyr和Thr修饰的多肽:我们提供单磷酸化和多磷酸化多肽服务,目前我们已经能够提供四个磷酸化位点修饰的多肽。2.5(6)-FAM,FITC,CY5,RhodamineB,PNA,EDNAS/dabcyl等荧光标记修饰的多肽:荧光标记修饰多肽技术是我们国肽生物的代表性多肽合成技术,我们的这项技术已经相当成熟。3.生物素Biotin,Lys(Biotin)修饰的多肽:生物素是维生素B2的组成部分,Biotin,Lys(Biotin)修饰的多肽也是客户经常定制的多肽。我们提供生物素修饰的多肽已经有将近100%的成功率。4.含有一对或多对二硫键修饰的多肽:二硫键在蛋白质的结构稳定中起到重要作用,目前我们已经能够为客户提供四对二硫键修饰的多肽。5.含有同位素C13,N15修饰的多肽:同位素标记的多肽主要应用于医学和生物学领域,通常价格较高,为了满足客户需要,我们接受微克级的同位素多肽定制。6.含有特殊氨基酸修饰的多肽:例如,D型氨基酸,氨基酸衍生物,脂肪族羧酸等等,都在我们接受的定制范围内。提供150个氨基酸以内的长肽合成服务多肽合成过程中,肽链过长时,经常会出现缺残基,氨基酸缩合困难等情况,基于这些现象,我们开发了三种有效提高反应成功率的方案:1. 微波合成法:对于合成过程中出现的一些难以缩合的氨基酸,我们采用微波法进行合成,该方法效果显著,并且大大缩短了反应时间。2. 片段合成法:当某些多肽用常规合成方法合成困难,我们也会采用将多肽中某一段的某几个氨基酸缩合之后作为一个整体缩合到肽链上去,这种方法也能够解决许多合成中存在的问题。3.酰肼合成法:酰肼法合成多肽的方法是将固相合成的 N末端Cys 多肽和 C末端多肽酰肼之间的化学选择性反应形成酰胺键而实现多肽的连接,该方法根据肽链中Cys的位置,将整条肽链分成多条序列分别合成,最终经过液相缩合反应得到目标肽,显著地提高了最终产物纯度,广泛适用于含有Cys的长链多肽的合成。国肽生物拥有成熟的长肽合成工艺,能够根据客户定制的多肽序列,快速有效地设计合成方案并迅速开始合成,更快更好的为客户提供所需的服务是我们不变的坚持。国肽生物主要提供:多肽合成、多肽定制、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、美容肽、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。详情请咨询国肽生物17730030462微信号Email:peptide50@bankpeptide.net[img=,690,143]https://ng1.17img.cn/bbsfiles/images/2021/02/202102031611349730_9244_3531468_3.jpg!w690x143.jpg[/img]
最近要测羰基指数,样品为PTMEG(聚四亚甲基醚二醇)是是四氢呋喃的聚合物。测它的羰基指数怎么弄?一般找那个峰为基准?现在只知道有文献上写羰基指数CI=Ac=o/Aref..... 求高人指点
有哪位老师检测过己烷(正己烷与环己烷等的混合物,出峰有6个成分)和甲基叔丁基醚的残留检测?我现在用了DB-1、DB-17、INNOWAX和DB624都没有分开,甲基叔丁基醚与己烷中的某一成分峰完全重合,请指点!谢谢!
急求甲基叔丁基醚中硫化物的测定方法:主要是用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定甲基叔丁基醚中硫醇,硫醚。要用什么样的色谱柱及有相关中外文献可查吗?
有谁知道叔丁基二甲基氯硅烷含量怎么测定啊?
实验室有麦可林 500ml/瓶的 甲基叔丁基醚 还有60瓶左右 一直没有使用 有需要的可以联系
请问有国产的叔丁基甲基醚吗?我们做实验需要用到这个试剂(T-BME),找了好几家试剂公司都说有进口的,而且还要14天的定货周期,全是2.5升的.不知道有没有国产试剂,小包装的.谢谢!
最近在做实验,需要计算手动校正因子。关于有效碳数的计算。想问问大家,异丙醇,甲基叔丁基醚,甲醛,乙醛,丙烯酸,叔丁醇的相对校正因子的计算。望大家给解答下,谢谢。
羰基镍 Nickel carbonyl CAS:13463-39-3[color=#ff0000]理化性质[/color]具有霉味的无色至淡黄色易挥发液体。分子式C4-Ni-O4。化学式 Ni(CO)4。分子量 170.73。相对密度 1.318(17℃)。熔点 -19.3℃。沸点 43℃。闪点 -20℃。自燃点 93.33℃。蒸气密度 5.95(50℃)。蒸气压 53.32kPa(400mmHg 25.8℃)。蒸气与空气混合物可燃下限 2% 。水中溶解度为0.018g/100ml 不溶于稀酸、稀碱 溶于乙醇、苯、氯仿、丙酮、四氯化碳、王水、乙醚、硝酸。液态羰基镍侵蚀某些塑料、橡胶、涂层。空气中氧化,与氧化剂反应生成一氧化碳和相应的盐。遇热、明火、氧化剂易燃。20℃时,它的蒸气在空气和氧气中的分压达到2.00 kPa(15 mmHg)时爆炸 液态羰基镍在60℃时爆炸。不能与硝酸、氯、溴、可燃性蒸气共存。[color=#ff0000]消防措施[/color][color=#0000ff][size=5][sup] [/sup][/size][/color]消防人员须穿戴全身防护服。用雾状水、泡沫、二氧化碳、干粉灭火。[color=#ff0000]储运须知[/color]包装标志:毒害品。包装方法:(I)类。高强度玻璃瓶充一氧化碳或其他不反应气体,气密封口,装在金属罐内,周围以惰性吸收材料衬填,外木箱或钢瓶装。储存条件:储存于阴凉、干燥、通风良好的仓库内。远离热源和火源。避光储存。仓库温度控制在28℃以下。搬运时轻装轻卸,防止容器破损。[color=#ff0000]泄漏处理[/color]切断一切火源,戴好防毒面具等全部防护用品。用不燃性分散剂制成的乳液刷洗。如无分散剂可用砂土吸收,倒至空旷地方掩埋。对污染地面用肥皂或洗涤剂刷洗,经稀释的污水放入废水系统。[color=#ff0000]接触机会[/color]羰基镍主要用于精炼镍、制造丙烯酸和甲基丙烯酸酯、有机合成的催化剂、作为钢和其他金属涂层、在冶金和电子工业中用于汽相扩散渗镀。当一氧化碳通入金属镍可形成不稳定的羰基镍。[color=#ff0000]侵入途径[/color]]主要经呼吸道吸入,也能经皮肤吸收。[color=#ff0000]毒理学简介[/color]人吸入TCLo: 7 mg/m3 LCLo: 30 ppm/30M。大鼠吸入LC50: 35 ppm/30M(244mg/m3)。小鼠吸入LC50: 67 mg/m3/30M。羰基镍为高毒物质。兔吸入浓度为 291mg/m^3 后 5分钟,发现镍在肺、血和肾的滞留量分别为 38.1 %、11.5%及 7.9%,肝内含量甚微。三天内约可随尿排出吸收镍的 62.2%。给大鼠 LD50 的剂量,经静脉、皮下、腹腔投毒后,24小时内脏器官肉眼检查无变化,第二天可见肺、肝脏肿大。肺部病变表现为肺水肿和灶性出血,肺血管周围有炎症细胞侵润,肺泡上皮细胞肥大和增生,肺泡壁增厚。肝脏为肝小叶中央中度淤血。中枢神经系统水肿,大脑半球毛细血管出血。约经二周后存活动物病理变化可趋好转。有人曾对一例接触羰基镍后13天急性中毒死亡的管道装配工人进行尸检。肺主要病变为肺实质由于成纤维细胞侵润而使很多区域硬变,只有很少量含有空气的软区。
问题:一个老产品,液相分析,流动相中用到甲基叔丁基醚,占流动相比例约20%,正常主峰出峰时间约25~27分钟,总分析时间60分钟,最近两年在夏天天气比较热时经常出现的问题是新配制好的流动相出峰时间明显延迟,有时候延迟至35分钟以后,补加少量甲基叔丁基醚会提前(当然这不符合规定,而且加多少量也没有规律)已排查措施:1、试剂品牌:该品牌试剂已使用多年,并且在其它季节没有问题,可以判断是天气炎热时才有这个问题2、温度:液相有柱温控制,液相室有空调(是新配制的流相相有问题,所以和使用过程挥发没有关系),试剂室及配制室没有空调,目前已将待用试剂放入冰柜,临用先取出来。3、人员:操作员工比较有经验,该流动相已配制无数次,可以排除人员的偶然偏差4、试剂质量:因问题只出现在天气炎热时,将“有问题”的甲基叔丁基醚检测气相纯度及水分,均未见异常5、因流动相及梯度略复杂,不适合在线混合。请高手多多指点~~谢谢~~
[color=#444444]如题,本实验室的GC-MC进样口温度为300度,对甲基苯甲酸叔丁酯的沸点也没查到,但至少都得200多度以上吧,甚至300度,比较难气化,不知道有谁做过没或者懂的帮我分析下,如果换成苯甲酸叔丁酯呢,沸点多少,能在GC-MC检测出来吗[/color][color=#444444]注:关键暂时没有对甲基苯甲酸叔丁基酯的标准物[/color]
各位老师,求一份做叔丁基甲基醚的翻译标准,U.SEPA8260D-2018这个标准的翻译版标准,看不懂英文。
本人需要大约3L左右的甲基叔丁基醚(分析纯),有用过或知道该联系方式的请告诉我好吗?谢谢我的邮箱:qqxlily0565@163.com
请问为什么甲基叔丁基醚能做为萃取剂,而且有取代乙醚成为萃取剂的趋势
求购分析纯的甲基叔丁基醚,最好是广州的厂家,联系邮箱:qqxlily0565@163.com
水质中测定甲基叔丁基醚 mtbe 大家有方法吗?
[color=#444444]分析类胡萝卜素类物质,查了几篇文献,都用到了[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]分析.文献的分析方法中用的都是离子阱色谱,流动相中除了甲醇都有甲基叔丁基醚.我准备根据文献的方法做[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]分析,可是联系了几个高校的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],不论质谱是不是离子阱都说流动相中不能用甲基叔丁基醚。[/color][color=#444444]难道文献的方法都是错误的?[/color][color=#444444]求助,哪个分析平台的质谱流动相可以有甲基叔丁基醚呀?[/color]
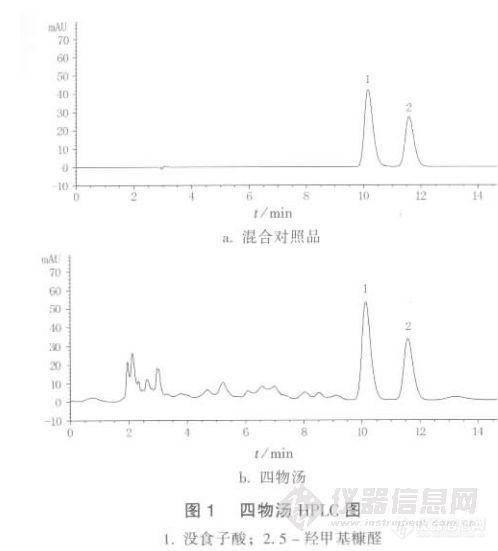
【作者】:雷 鹏 ,李 媛,李新中,黄 义【摘要】:目的 建立 Q7W- 法同时测定四物汤中没食子酸、D E 羟甲基糠醛含量的方法,并比较四物汤传统饮片汤剂与配方颗粒汤剂中没食子酸、D E 羟甲基糠醛的含量。方法 1(&8*+0(, -G# 色谱柱 H !B " 88 X ;D 88,D !8J ;流动相:甲醇 E
EN 14362-1 2012标准上说用甲基叔丁基醚经硅藻土柱萃取还原后的样品溶液后,用旋转蒸发仪将甲基叔丁基醚浓缩至1ml左右(不能蒸干,蒸干可能会使芳香胺损失),然后用低流速的惰性气体吹干。我的问题是把甲基叔丁基醚蒸发干会可能使芳香胺损失,那用惰性气体把甲基叔丁基醚吹干就不会有损失吗?求解答?
最近想要用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]测类胡萝卜素,查阅文献发现流动相要用到甲基叔丁基醚,工程师不建议用MTBE做流动相,请问论文里一般都是怎么做的呀?
各位大侠,本部门在开展自来水106项中二氯乙酸,三氯乙酸任务时,使用甲基叔丁基醚4mL萃取酸化后(pH2)40mL水样中的二氯乙酸,三氯乙酸,后取用其中2ml甲基叔丁基醚与2mL(10%硫酸酸化的)甲醇于45℃水浴中反应降温保护卤乙酸甲酯后,使用7mL150g/L的硫酸钠水溶液对其进行萃取问题来了,硫酸钠溶液萃取前,有机溶剂剩余4mL(甲醇2mL,甲基叔丁基醚2mL),萃取之后却发现有机相大大缩水,仅剩余1mL以下萃取的目的是为了除去有机相中的甲醇,保留甲基叔丁基醚,结果发现,大量甲基叔丁基醚消失,怀疑形成甲醇-甲基叔丁基醚-水共溶,因此,做了验证试验,加入2mL甲醇,2mL甲基叔丁基醚于分液漏斗,加入7mL150g/L硫酸钠溶液,摇晃后静置15min,期间不加热,不加酸,结果留下的有机相也仅为1mL。请教诸位达人,中是否可能形成甲醇-甲基叔丁基醚-水共溶,诸位在卤乙酸的gc检测预处理过程中采用何种方法??急求,在线等
最近在用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url],对选用的溶剂甲基叔丁基醚存在疑惑,请问它能作为[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]的溶剂吗,具体原因,万分感谢!
大家说说这两种醚萃取的特点,为什么有的检测项目要用甲基叔丁基醚呢,味道好大,熏死了
气相色谱质谱法测定土壤中甲基叔丁基醚1.方法概述1.1方法原理样品中的挥发性有机物经高纯氦气(或氮气)吹扫后吸附于捕集管中,将捕集管加热并以高纯氦气反吹,被热脱附出来的组分经气相色谱分离后,用质谱仪进行检测。通过与待测目标化合物保留时间和标准质谱图或特征离子相比较进行定性,内标法定量。1.2方法依据HJ 605—2011土壤和沉积物 挥发性有机物的测定 吹扫捕集/气相色谱-质谱法1.3方法适用范围本标准规定了测定土壤和沉积物中挥发性有机物的吹扫捕集/气相色谱-质谱法。本标准适用于土壤和沉积物中65种挥发性有机物的测定。若通过验证本标准也可适用于其他挥发性有机物的测定。当样品量为5g,用标准四极杆质谱进行全扫描分析时,目标物的方法检出限为0.2~3.2μg/kg,测定下限为0.8~12.8μg/kg。2.试剂和材料2.1空白试剂水:二次蒸馏水或通过纯水设备制备的水,使用前需经过空白检验,确认在目标化合物的保留时间区内无干扰峰出现或目标化合物浓度低于方法检出限。 2.2甲醇(CH4O):农残级,使用前需通过检验,确认无目标化合物或目标化合物浓度低于方法检出限。2.3内标标准储备液:2000mg/L于甲醇中,溯源号:120004-02,批号:396190,品牌o2si。2.4替代物储备溶液:2000mg/L于甲醇中,溯源号:120005-01,批号:394601,品牌o2si。2.5甲基叔丁基醚储备液:2000mg/L于甲醇中,溯源号:CDAA-S-620126-AE-1ml,批号:K5580060,品牌安谱。2.6载气:氮气,纯度99.999%,用净化管净化。2.7载气:氦气,纯度99.999%,用净化管净化。3.仪器和设备注:本标准除特殊说明,分析时均使用符合国家标准A级标准的玻璃量器。3.1气相色谱/质谱仪:安捷伦7890B-5977B,带有EI源,编号为QNJC/FX-083。3.2吹扫捕集样品浓缩仪:美国OI Analytical Eclipse 4760/4100,编号为QNJC/FX-154。3.3低温恒温槽:DC-2006,编号为QNJC/FX-152。3.4分析天平:精度为0.01g,编号为QNJC/FX-023。3.5毛细管色谱柱:安捷伦安捷伦DB-624,60m×250μm×1.4μm。3.6容量瓶:10ml,精度为0.02ml。3.7微量注射器:10μL、25μL、100μL、500μL。3.8安捷伦进样瓶:2ml。3.9棕色VOA瓶:40 ml棕色玻璃瓶,具聚四氟乙烯硅胶垫螺旋盖。4.样品与现场质控样采集4.1样品采集土壤和沉积物样品的采集分别参照HJ/T166和GB17378.3的相关规定。可在采样现场使用用于挥发性有机物测定的便携式仪器对样品进行目标物含量高低的初筛。所有样品均应至少采集 3 份平行样品,并用60ml样品瓶(或大于60ml其他规格的样品瓶)另外采集一份样品,用于测定高含量样品中的挥发性有机物和样品含水率。4.1.1手工进样方式的采样方法本采样方法适用于无自动进样器的吹扫捕集装置。用铁铲或药勺将样品尽快采集至60ml样品瓶(或大于60ml其他规格的样品瓶)中,并尽量填满。快速清除掉样品瓶螺纹及外表面上黏附的样品,密封样品瓶。4.1.2 自动进样方式的采样方法本采样方法适用于带有自动进样器的吹扫捕集装置。采样前在每个40ml棕色样品瓶中放一个清洁的磁力搅拌棒,密封,贴标签并称重(精确至0.01g),记录其重量并在标签上注明。采样时,用采样器采集适量样品到样品瓶中,快速清除掉样品瓶螺纹及外表面上黏附的样品,密封样品瓶。4.2现场质控样采集。4.2.1全程序空白样采样前在实验室将空白试剂水放入样品瓶中密封,将其带到采样现场。与采样的样品瓶同时开盖和密封,随样品运回实验室,按与样品相同的分析步骤进行处理和测定,用于检查样品采集到分析全过程是否受到污染。4.2.2运输空白样采样前在实验室将空白试剂水放入样品瓶中密封,将其带到采样现场。采样时其瓶盖一直处于密封状态,随样品运回实验室,按与样品相同的分析步骤进行处理和测定,用于检查样品运输过程中是否受到污染。4.2.3现场平行样跟随土样采集时在同一个采样点在同一时间、同样方法采集双份样品作为现场平行样。现场平行样的测定分析同其余样品。当每批次样品量≤20个时,应至少采集5%现场平行样。4.2.4现场加标样(无)5.样品运输和保存样品采集后应冷藏避光运输,运回实验室后应尽快分析。实验室内样品存放区域应无有机物干扰,在4℃以下保存时间为7 d。6.样品分析6.1样品含水率的测定取5g(精确至0.01 g)样品在(105±5)℃下干燥至少6 h,以烘干前后样品质量的差值除以烘干前样品的质量再乘以100,计算样品含水率w(%),精确至0.1%。6.2样品前处理6.2.1低含量样品的测定若初步判定样品中挥发性有机物含量小于200 μg/kg时,用5g样品直接测定;初步判定含量为200~ 1000μg/kg时,用1g样品直接测定。若吹扫捕集装置带有自动进样器时,将样品瓶轻轻摇动,确认样品瓶中的样品能够自由移动,称量并记录样品瓶重量(精确至0.01g)。用气密性注射器量取10.0ml空白试剂水,用微量注射器分别量取 25.0μL浓度为10mg/L的内标标准溶液和25.0μL浓度为10mg/L的替代物标准溶液加入样品瓶中,按照仪器参考条件(6.5)进行测定。6.2.2 高含量样品的测定对于初步判定目标物含量大于1000μg/kg的样品,从样品瓶中取5g左右样品于预先称重的40ml 无色样品瓶中,称重(精确至0.01g),然后迅速加入10.0ml甲醇,盖好瓶盖并振摇2min。静置沉降后,用微量注射器分别量取10.0~100μL提取液、25.0μL浓度为10mg/L的内标标准溶液和25.0μL浓度为10mg/L的替代物标准溶液至用气密性注射器量取的10.0ml空白试剂水作为试料,放入40ml样品瓶中(若无自动进样器,则直接放入吹扫管中),按照仪器参考条件(6.5)进行测定。6.3空白样品的制备称取5g石英砂并且加入10ml空白试剂水作为实验室分析空白样品,连同全程序空白样和运输空白样参照(6.2)中所示前处理过程进行分析测定。6.4曲线的制备6.4.1标准使用溶液的配制6.4.1.1替代物使用液:用微量进样针分别准确移取50μL浓度为2000mg/L的替代物储备液于10ml容量瓶中,用甲醇定容至刻度线得到浓度为10mg/L的使用液。6.4.1.2内标物使用液:用微量进样针准确移取50μL浓度为2000mg/L的内标物储备液于10ml容量瓶中,用甲醇定容至刻度线得到浓度为10mg/L的内标物使用液。6.4.1.3甲基叔丁基醚使用液:移取50μL浓度为2000mg/L的标液于10ml容量瓶中,用甲醇定容至刻度线得到浓度为10mg/L的混标使用液。6.4.2校准曲线的绘制6.4.2.1校准曲线系列的配制分别移取替代物使用液(10mg/L)、内标物使用液(10mg/L)和甲基叔丁基醚标液(10mg/L)至装有10ml空白试剂水的棕色VOA瓶中,取标体积如表1所示,加标以后立即用带有聚四氟乙烯垫片的盖子将棕色VOA瓶盖紧并放入自动吹扫捕集浓缩仪中进行测定。表1 土壤挥发性有机物标准曲线配制表名称名称浓度(mg/L)取标体积(μL)定容体积(ml)最终浓度(μg/L)STD-1替代物使用液105105内标使用液1025甲基叔丁基醚105STD-2替代物使用液10101010内标使用液1025甲基叔丁基醚1010STD-3替代物使用液10251025内标使用液1025甲基叔丁基醚1025STD-4替代物使用液10501050内标使用液1025甲基叔丁基醚1050STD-5替代物使用液1010010100内标使用液1025甲基叔丁基醚101006.4.2.2用平均相对响应因子绘制校准曲线校准系列第 i 点中目标物(或替代物)的相对响应因子(RRFi),按照(1)式进行计算:RRFi= ……………………………………………………......(1)式中:RRFi ——标准系列中第 i 点目标物(或替代物)的相对响应因子;AIsi ——标准系列中第 i 点目标物(或替代物)相对应内标定量离子的响应值;Ai ——标准系列中第 i 点目标物(或替代物)的定量离子的响应值;CIS ——标准系列中内标的质量浓度,μg/L;Ci ——标准系列中第 i 点目标物(或替代物)的质量浓度,μg/L。目标物(或替代物)的平均相对响应因子,按照式(2)计算:https://ng1.17img.cn/bbsfiles/images/2021/10/202110081402244827_4731_3960693_3.png………………………………………………….............(2)式中:——目标物(或替代物)的平均相对响应因子; RRFi ——标准系列中第i点目标物(或替代物)的相对响应因子; n ——标准系列点数。RRF的标准偏差(SD),按照式(3)进行计算:https://ng1.17img.cn/bbsfiles/images/2021/10/202110081402243491_5129_3960693_3.png………………………………………………….............(3)RRF的相对标准偏差(RSD),按照式(4)进行计算:RSD= 100% …………………………………………………...............(4)任一化合物和替代物的平均响应因子的相对标准偏差不能超过20%。如果初始校准中多于10%的化合物的相对标准偏差超过20%,并且线性相关系数无法达到最小限值(0.99),那么该色谱系统被视为不够精确、不能进行分析,需要重新配制校准曲线进行校准。6.5样品与空白样的测定样品与空白样品的前处理及分析步骤按照(6.2)中所示进行。同时本方法使用的色谱及吹扫捕集分析条件如下所示:(a)气相色谱参考条件:色 谱 柱:安捷伦DB-624(60m×250μm×1.4μm)。程序升温:40℃(2min)→5℃/min→120℃→10℃/min→230℃(4min)。进样口温度:230℃。柱(氦气)流量:1.0 ml/min。分流比:8:1。 (b)质谱参考条件:扫描方式:全扫描(SCAN)。扫描范围:35~550amu。离子源温度:230℃。四级杆温度:150℃。传输线温度:230℃。离子化能量:70eV。溶剂延迟:2min。(c)吹扫捕集条件吹扫温度:40℃。吹扫时间:11min。预脱附温度:180℃。脱附温度:190℃。烘烤温度:210℃。烘烤时间:10min。(d)仪器性能核查在仪器使用前、样品分析前及每运行24 h,气相色谱质谱仪系统必须进行仪器性能检查。取调谐标准溶液BFB加入到10ml空白试剂水中,通过吹扫捕集装置进样,用GC/MS进行分析,得到的BFB关键离子丰度应满足下表 2 的规定标准。否则需对质谱仪的一些参数进行调整或清洗离子源。表2 4-溴氟苯(BFB)关键离子及丰度标准质荷比(m/z)丰度标准质量离子(m/z)丰度标准50质量95的8%~40%174大于质量95的50%75质量95的30%~80%175质量174的5%~9%95基峰,100%相对丰度176质量174的93%~101%96质量95的5%~9%177质量176的5%~9%173小于质量174的2%--6.6 结果计算与表示6.6.1定性分析对于每一个目标化合物,应使用标准溶液或通过校准曲线经过多次进样建立保留时间窗口,保留时间窗口为±3倍的保留时间标准偏差,样品中目标化合物的保留时间应在保留时间的窗口内。对于全扫描方式,目标化合物在标准质谱图中的丰度高于30%的所有离子应在样品质谱图中存在,而且样品质谱图中的相对丰度与标准质谱图中的相对丰度的绝对值偏差应小于20%。例如,当一个离子在标准质谱图中的相对丰度为30%,则该离子在样品质谱图中的丰度应在10%~50%之间。对于某些化合物,一些特殊的离子如分子离子峰,如果其相对丰度低于30%,也应该作为判别化合物的依据。如果实际样品存在明显的背景干扰,则在比较时应扣除背景影响。在本方法测定条件下的挥发性有机物总离子流图如下图1所示:https://ng1.17img.cn/bbsfiles/images/2021/10/202110081403509740_1487_3960693_3.png!w690x327.jpg" style="max-width: 100%;max-height: 100%;1、甲基叔丁基醚; 2、二溴氟甲烷(替代物); 3、氟苯(内标); 4、甲苯-D8(替代物); 5、氯苯-D5(内标); 6、4-溴氟苯(替代物); 7、1,4-二氯苯-D4(内标); 图1 挥发性有机物的总离子流图6.6.2定量分析当目标物(或替代物)用平均相对响应因子进行校准时,试料中目标物的质量浓度CX按照式(5)CX= ………………………………………...………...............(5)式中:CX ——样品中目标化合物的质量浓度,μg/L;AX ——目标化合物定量离子的响应值;AIS ——目标化合物相对应内标定量离子的响应值;CIS ——内标物的质量浓度,μg/L;——目标化合物的平均相对响应因子;当目标物采用线性或非线性校准曲线进行校准时,试料中目标物质量浓度CX通过相应的校准曲线计算。1)对于低含量样品,样品中目标物的含量(μg/kg)按照式(6)进行计算:ω=………………………………………..……...............(6)式中:ω——样品中目标物的含量,μg/kg;5 ——试料体积,ml;CX ——试料中目标物的质量浓度,μg/L;w ——样品的含水率,%;m ——样品量,g2)对于高含量样品,样品中目标物的含量(μg/kg)按照式(7)进行计算:ω=……………………………………………...............(7)式中:ω——样品中目标物的含量,μg/kg;5 ——试料体积,ml;CX ——试料中目标物的质量浓度,μg/L;VC——提取液体积,ml;w ——样品的含水率,%;Vs——用于吹扫的提取液体积,ml;m ——样品量,g;k——提取液稀释倍数。注:若样品含水率大于10%时,提取液体积VC应为甲醇与样品中水的体积之和;若样品含水率小于等于10%,提取液体积VC为10 ml。6.6.3 结果表示当测定结果小于100μg/kg时,保留小数点后1位;当测定结果大于等于100μg/kg时,保留3位有效数字。在使用本标准规定的毛细管柱时,间二甲苯和对二甲苯的测定结果为两者之和。7.质量保证和质量控制7.1检出限测定对5g石英砂进行体积为2μL浓度为10mg/L的加标实验,前处理步骤同(6.1~6.2)中所示,平行测定7次后测得的检出限值如下表1-1和1-2:表1-1 土壤中挥发性有机物检出限测定值(t值为3.143) 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%检出限MDL (μg/kg)甲基叔丁基醚4.84.24.24.04.04.24.04.20.36.71.0结论:经过7次平行测定得出土壤在本方法的检出限为1.0μg/kg,满足标准规定的检出限值(0.2μg/kg~3.2μg/kg)。 表1-2 沉积物中挥发性有机物检出限测定值(t值为3.143) 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%检出限MDL (μg/kg)甲基叔丁基醚4.23.83.83.63.43.64.03.80.37.11.0结论:经过7次平行测定得出沉积物在本方法的检出限为1.0μg/kg,满足标准规定的检出限值(0.2μg/kg~3.2μg/kg)7.2测定下限本方法以4倍检出限来作为测定下限,最终的测定下限值如下表2-1和2-2中所示:表2-1 土壤方法验证测定下限值 化合物验证测定下限(μg/kg)方法测定下限(μg/kg)甲基叔丁基醚1.04.0表2-2 沉积物方法验证测定下限值 化合物验证测定下限(μg/kg)方法测定下限(μg/kg)甲基叔丁基醚1.04.0结论:根据表中所示结果得出实验得出的测定下限都满足方法HJ605-2011中规定的数值。7.3精密度测定7.3.1现场平行对土壤和沉积物分别进行2次平行测定,测定的结果如下表3和表4所示:表3 土壤样品平行性测定结果(单号:21MN0009) 化合物12平均值(μg/kg)相对偏差%甲基叔丁基醚47.553.350.45.8表4 沉积物样品平行性测定结果(单号:21MN0010) 化合物12平均值(μg/kg)相对偏差%甲基叔丁基醚54.149.852.04.1结论:对土壤和沉积物样品分别进行2次平行测定,平行样品之间的相对偏差均满足≤20%的要求。7.3.2分析平行对5g石英砂模拟土壤基质分别进行5μL(10mg/L)、25μL(10mg/L)、100μL(10mg/L)低、中、高三种不同浓度的加标实验,然后加入10ml实验室空白水样,前处理步骤同(6.1~6.2)中所示,三种浓度各平行测定7次后测得的检出限值如表5-1~表7-1所示:表5-1 曲线低浓度点加标实验测定值 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%甲基叔丁基醚8.49.08.88.49.88.69.08.90.55.5表6-1 曲线中间浓度点加标实验测定值 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%甲基叔丁基醚56.050.251.654.257.853.252.253.62.64.9表7-1 曲线高浓度点加标实验测定值 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%甲基叔丁基醚208224197.623622022422421912.55.7结论:从上述三种浓度加标实验测定值得出甲基叔丁基醚进行低、中、高三种不同浓度加标测定的精密度值(RSD%)为5.5%、4.9%、5.7%,满足相对标准偏差(RSD%)≤15%的要求。对5g沉积物基质分别进行5μL(10mg/L)、25μL(10mg/L)、100μL(10mg/L)低、中、高三种不同浓度的加标实验,然后加入10ml实验室空白水样,前处理步骤同(6.1~6.2)中所示,三种浓度各平行测定7次后测得的检出限值如表5-2~表7-2所示:表5-2 曲线低浓度点加标实验测定值 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%甲基叔丁基醚8.08.48.28.08.08.48.68.20.23.0表6-2 曲线中间浓度点加标实验测定值 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%甲基叔丁基醚57.051.250.451.652.658.255.453.83.15.7表7-2 曲线高浓度点加标实验测定值 化合物1234567平均值(μg/kg)标准偏差SD(μg/kg)相对标准偏差RSD%甲基叔丁基醚2322142282242342302362287.43.3结论:从上述三种浓度加标实验测定值得出甲基叔丁基醚进行低、中、高三种不同浓度加标测定的精密度值(RSD%)为3.0%、5.7%、3.3%,满足相对标准偏差(RSD%)≤15%的要求。7.4准确度测定7.4.1质控样(无)7.4.2加标回收率对5g石英砂和沉积物进行基质加标进行回收率测定,加标体积为25μL,加标浓度为10mg/L,然后按照(6.1~6.2)中所示的前处理步骤进行分析测定,最终的加标结果如下表8-1和8-2所示:表8-1 土壤挥发性有机物加标回收率测定值 化合物1234567平均值(μg/kg)加标量(μg/kg)加标回收率%甲基叔丁基醚56.050.251.654.257.853.252.253.650.0107表8-2 土壤挥发性有机物加标回收率测定值 化合物1234567平均值(μg/kg)加标量(μg/kg)加标回收率%甲基叔丁基醚57.051.250.451.652.658.255.453.850.0108结论:对5g石英砂和沉积物样品分别进行甲基叔丁基醚加标,测得的回收率分别为107%和108%,满足加标回收率70%~130%的要求。7.5空白样测定7.5.1全程序空白采样前在实验室将空白试剂水放入样品瓶中密封,将其带到采样现场。与采样的样品瓶同时开盖和密封,随样品运回实验室,按与样品相同的分析步骤进行处理和测定,测定结果如下表9所示:表9 全程序空白测定结果 化合物测定值(μg/kg)检出限(μg/kg)甲基叔丁基醚1.01.0结论:经过测定得出全程序空白的测定结果满足小于方法检出限值的要求。7.5.2运输空白采样前在实验室将空白试剂水放入样品瓶中密封,将其带到采样现场。采样时其瓶盖一直处于密封状态,随样品运回实验室,按与样品相同的分析步骤进行处理和测定。对运输空白的测定结果如下表10所示:表10 现场空白测定结果 化合物测定值(μg/kg)检出限(μg/kg)甲基叔丁基醚1.01.0结论:经过测定得出运输空白的测定结果满足小于方法检出限值的要求。7.5.3分析空白取5g石英砂加入10ml实验室空白水样,按照(6.1)中步骤进行全程序测定,分析空白的测定结果如下表11所示:表11 分析空白测定结果 化合物测定值(μg/kg)检出限(μg/kg)甲基叔丁基醚1.01.0结论:经过测定得出分析空白的测定结果满足小于方法检出限值的要求。7.6曲线校准 用微量进样针分别移取替代物使用液(10mg/L)、内标物使用液(10mg/L)和甲基叔丁基醚标液(10mg/L)至装有10ml空白试剂水的棕色VOA瓶中,取标体积如表1所示,配制成浓度为5μg/L、10μg/L、25μg/L、50μg/L、100μg/L的校准曲线系列。最后以浓度比为横坐标,响应比为纵坐标绘制标准曲线。所得的曲线信息如下表12所示:表12 土壤挥发性有机物校准曲线 化合物校正曲线方程式响应因子(RSD%)甲基叔丁基醚y=1.073x9.532结论:化合物甲基叔丁基醚的校准曲线均满足方法响应因子相对标准偏差≤20%的要求。8.结论 (1)对5g石英砂和沉积物样品分别进行体积为2μL浓度为10mg/L的加标实验,经过7次平行测定得出土壤和沉积物在本方法的检出限为1.0μg/kg,满足标准规定的检出限值(0.2μg/kg~3.2μg/kg)。(2)本方法以4倍检出限来作为测定下限,实验得出的测定下限都满足方法HJ605-2011中规定的数值。(3)对土壤和沉积物样品分别进行2次平行测定,平行样品之间的相对偏差均满足≤20%的要求。(4)对5g石英砂分别进行5μL(10mg/L)、25μL(10mg/L)、100μL(10mg/L)低、中、高三种不同浓度的加标实验,然后加入10ml实验室空白水样,从上述三种浓度加标实验测定值得出土壤挥发性有机物中甲基叔丁基醚的精密度(RSD%)为5.5%、4.9%、5.7%,满足相对标准偏差(RSD%)≤15%的要求;对5g沉积物基质分别进行5μL(10mg/L)、25μL(10mg/L)、100μL(10mg/L)低、中、高三种不同浓度的加标实验,然后加入10ml实验室空白水样,从上述三种浓度加标实验测定值得出沉积物挥发性有机物中甲基叔丁基醚的精密度(RSD%)为3.0%、5.7%、3.3%,满足相对标准偏差(RSD%)≤15%的要求。(5)对5g石英砂和沉积物分别进行基质加标进行回收率测定,加标体积为25μL,加标浓度为10mg/L,测得的平均回收率分别为107%和108%,满足加标回收率70%~130%的要求。(6)采样前在实验室将空白试剂水放入样品瓶中密封,将其带到采样现场。与采样的样品瓶同时开盖和密封,随样品运回实验室,按与样品相同的分析步骤进行处理和测定,经过测定得出全程序空白的测定结果满足小于方法检出限值的要求。(7)采样前在实验室将空白试剂水放入样品瓶中密封,将其带到采样现场。采样时其瓶盖一直处于密封状态,随样品运回实验室,按与样品相同的分析步骤进行处理和测定,经过测定得出运输空白的测定结果满足小于方法检出限值的要求。(8)取5g石英砂加入10ml实验室空白水样,经过测定得出分析空白的测定结果满足小于方法检出限值的要求。(9)用微量进样针分别移取替代物使用液(10mg/L)、内标物使用液(10mg/L)和甲基叔丁基醚标液(10mg/L)至装有10ml空白试剂水的棕色VOA瓶中,取标体积如表1所示,配制成浓度为5μg/L、10μg/L、25μg/L、50μg/L、100μg/L的校准曲线系列。最后以浓度比为横坐标,响应比为纵坐标绘制标准曲线。化合物甲基叔丁基醚的校准曲线满足方法响应因子相对标准偏差≤20%的要求。综上所述,本检测中心具备对化合物甲基叔丁基醚使用HJ 605-2011土壤和沉积物 挥发性有机物的测定 吹扫捕集/气相色谱-质谱法的检测能力。9.注意事项9.1仪器分析过程中的主要干扰来自样品的交叉污染。当分析了高浓度样品后应插一针空白,再分析下一个样品。9.2在实验过程中应穿着实验服,佩戴好实验防护用具,如手套、护目镜等。9.3当测定结果小于100μg/kg时,保留小数点后1位;当测定结果大于等于100μg/kg时,保留3位有效数字。在使用本标准规定的毛细管柱时,间二甲苯和对二甲苯的测定结果为两者之和。9.4实验过程中使用的标品及试剂对人体有害,应避免误食及接触皮肤,如沾染皮肤应及时用大量水冲洗,如误食应及时就医。9.5在使用玻璃器皿时应注意被玻璃划伤。9.6实验过程中产生的废弃物应注意收集,交由有资质的单位进行处理。9.7仪器进样口、柱箱和检测器等部件具有高温,在日常维护时应先对仪器进行降温,并且佩戴隔热手套防止烫伤。如不慎烫伤应及时擦拭烫伤膏,严重时需及时就医。9.8主要污染来自溶剂、试剂、不纯的惰性吹扫气体、玻璃器皿和其他样品处理设备。应使用纯化后的溶剂、试剂和惰性吹扫气体,样品贮存和分析时应尽量避免实验室中其他溶剂的污染,玻璃器皿和其他样品处理设备应清洗干净,不应使用非聚四氟乙烯密封垫圈、塑料管或橡胶组分的流量控制器,气相色谱载气管线及吹扫气管线应是不锈钢管或铜管,实验室分析人员的衣物不应有溶剂污染,特别是二氯甲烷污染。9.9每次分析样品前检查洗针瓶内空白试剂水是否用尽,并且检查废液瓶是否已满,及时倾倒废液避免溢出。9.10若样品中含有大量水溶性物质、悬浮物、高沸点有机化合物或高含量有机化合物,在分析完后需用肥皂水和空白试剂水清洗吹扫装置和进样针,然后在烘箱中105℃烘干。9.11若样品中有些高沸点有机化合物被吹脱出来,它们将在目标物之后流出色谱柱。在程序升温完成后,气相色谱应有烘烤时间确保高沸点有机化合物流出色谱柱。9.12酮类物质的吹扫温度升至 80 ℃,吹扫捕集效率和回收率可明显提高。9.13当分析空白试验样品时发现苯和苯乙烯出现异常高值,表明捕集肼中Tenax填料可能变质失效,需进行确认,必要时需更换捕集管。9.14在测定高含量样品时,若样品含水率大于10%时,提取液体积VC应为甲醇与样品中水的体积之和;若样品含水率小于等于10%,提取液体积VC为10ml。
求购叔丁基甲基醚色谱纯以及甲醛标液甲醛标液最好是100mg/l,20ml的那种环境标准物质,有的站内信~







 我要推广仪器
我要推广仪器
 下载APP
下载APP