離子源卸調後用甲醇洗後發現全氟三丁氨質譜圖中的770和866太低,高度接近峰底,遠遠沒有背景的119高,測試10溴deca出現上面相似 問題600以上峰都很低,不知道是離子遠電壓問題還是檢測器問題,燈絲也換過還是如此,求教大俠。!!
最近做样品中羰基化合物需要用到无醛甲醇,不想精制太麻烦,网上找了一下没找到,有没有做类似项目的给推荐一下用着不错的牌子,先谢了。
使用气象色谱仪测量甲醇发动机尾气排放中甲醇、乙醇、甲醛、乙醛含量,浓度在0到100ppm之间。请问应该使用什么类型的填充柱来分离呢?谢谢。
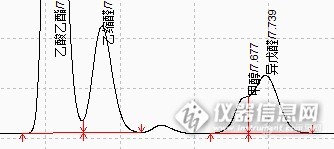
http://ng1.17img.cn/bbsfiles/images/2013/09/201309281119_468038_2451684_3.png乙缩醛和甲醇之间有一个峰,不知道是什么物质
你好请问你们做甲醇与乙醛的分离度是多少 我之前用DB-624柱子30M长的甲醇与乙醛分离度2.0,作了几次后,就分不开了 请问这是什么原因
分析白酒时乙醛和甲醇时间挨的近,出了乙醛峰很好,但是甲醇就不行了,又矮又长,怎么办啊?调节什么可以[em09501]
我们这边[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]是Angilent7890,我做乙醇挥发性物质时,试剂配制用了乙醛,为什么乙醛的出峰时间和甲醇(乙醇里面的杂质)一样,是不是我的操作有问题啊?请高手解答,谢谢。[color=#DC143C]被测溶液:取无水甲醇50µ l与乙醛50µ l,加96%乙醇稀释至50.0ml。取该溶液100µ l,加96%乙醇稀释至10.0ml。[/color]色谱柱:熔融石英,长:30m,内径:0.32mm,固定相:聚[(氰丙基)(苯基)][二甲基]硅氧烷R (膜厚1.8µ m)。载气:色谱用氦气。线速度:35cm/s。检测器:氢焰离子化检测器。柱温:0-12min:40℃,12-32min:40→240℃,32-42min:240℃。进样口温度:200℃。检测器温度:280℃。进样量:1µ l。这是欧洲药典的要求,既然药典这样写的,甲醇和乙醛肯定能分开出峰的,但是我按这要求做过后,两个峰分不开。为什么?注:我用的仪器是Agilent [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]7890,但柱子是国产的,那是不是柱子的问题呢?
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测器为FID检测器[/color][color=#444444]用色谱柱DB-624做时,出峰顺序为乙醛、甲醇、乙醇(这个是肯定的)[/color][color=#444444]用色谱柱RTX-5做时,出峰顺序为甲醇、乙醇、乙醛 (这是疑似)[/color][color=#444444]求教当用RTX-5做样时,这三个成分的出峰顺序,[/color][color=#444444]用RTX-5 做过的大哥,指点一下[/color]
按药典方法做,db-624色谱柱,0.25mm*1.4um,流速1ml,40度维持12分钟,每分钟10度升至240维持10分钟,分流5:1,出来的图如下,大神可否给看 看该怎么办?乙醛和无水甲醇峰都这么小吗?(图一)另一张是无水甲醇的(图二),如果不分流,无水甲醇和乙醇就连一起了(图三)求大神帮忙?[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251606373224_254_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251606376114_8345_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251606373624_9954_3443683_3.jpeg[/img]
我们这边[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]是Angilent7890,我做乙醇挥发性物质时,试剂配制用了乙醛,为什么乙醛的出峰时间和甲醇(乙醇里面的杂质)一样,是不是我的操作有问题啊?请高手解答,谢谢。被测溶液:取无水甲醇50µ l与乙醛50µ l,加96%乙醇稀释至50.0ml。取该溶液100µ l,加96%乙醇稀释至10.0ml。色谱柱:熔融石英,长:30m,内径:0.32mm,固定相:聚[(氰丙基)(苯基)][二甲基]硅氧烷R (膜厚1.8µ m)。载气:色谱用氦气。线速度:35cm/s。检测器:氢焰离子化检测器。柱温:0-12min:40℃,12-32min:40→240℃,32-42min:240℃。进样口温度:200℃。检测器温度:280℃。进样量:1µ l。这是欧洲药典的要求,既然药典这样写的,甲醇和乙醛肯定能分开出峰的,但是我按这要求做过后,两个峰分不开。为什么?注:我用的仪器是Agilent [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]7890,但柱子是国产的,那是不是柱子的问题呢?
小弟刚开始用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],请问我想检测进口葡萄酒、蒸馏酒中甲醇、总酸、总酯、总醛用哪种色谱柱效果好。
我用的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]器是安捷伦的7820A型,主要是是做无水乙醇挥发杂质的分析。 刚用这根柱子做的时候,乙醛和无水甲醇峰的分离度能达到2左右!(药典规定大于1.5)后来对仪器啊,色谱柱都不怎么熟悉,就又用这柱子做了一种有机胺化合物的含量。结果后来再做无水乙醇的时候,两个峰之间分离度就不够了,换了根新柱子也不行。而且现在的甲醇峰还有拖尾的现象,不知道是怎么回事。色谱柱:DB-624毛细管柱分离度:20:1升温程序: 时间 min 温度 ℃ 色谱柱 0---12 40 12---32 40到240 32---42 240 进样部位 -- 200 检测器 -- 280
做乙醇挥发性杂质测定时,按药典方法40度维持12分钟,每分钟10度升至240度,维持10分钟,使用db624柱子,0.25mm*1.4um,流速1ml,分流比5:1时,乙醛和无水甲醇峰特别小,峰形也不好(图一),单进无水甲醇的峰拖尾(图二),苯没出来,如果采取不分流进样,无水甲醇和乙醇峰就出一起了,大家做的图是什么样的,请大神帮忙看看怎么办?[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251637117241_6078_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251637118111_1179_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251637120861_5343_3443683_3.jpeg[/img]

化学试剂甲醇是以工业甲醇为原料,通过提纯而得。作者在徐州化工厂试剂分厂负责技术期间,利用传质效率低的闲置精馏塔,采用新工艺生产分析纯甲醇。试验证明:用化学试剂生产厂家通常采用的以氢氧化钠为化学处理剂改善甲醇中的“酸度”技术指标方法,不能使用传质效率较低的精馏塔把工业甲醇提纯至分析纯标准 而新工艺以适量的高锰酸钾和浓硫酸作化学催化剂,用传质效率较低的精馏塔能生产出分析纯甲醇。1提纯原理1.1 原料质量选用市场上供应的工业二级品甲醇,按参考文献所示方法对原料进行化验,结果见表1。[img]http://ng1.17img.cn/bbsfiles/images/2008/12/200812270838_126484_1643419_3.jpg[/img]由表1得知,工业甲醇中的蒸发残渣、水分、酸度、碱度、易炭化物质、羰基化合物、还原高锰酸钾等几项技术指标不符合分析纯标准。工业甲醇杂质的组成比较复杂,采用光谱分析和色谱—质谱分析,已发现40余种不同类型的有机含氧化合物,如C2~C6的醇、醛、酮、醚、甲醛缩二甲醇等。相对于甲醇的沸点(64.7℃),高沸点杂质可用精馏方法除去,低沸点杂质可通过在塔釜中加化学处理剂,与之反应生成高沸点的杂质精馏除去,以确保甲醇的质量符合分析纯标准。 化学试剂生产厂家一般利用盐酸羟胺与甲醇中的醛与酮杂质反应生成固体肟的原理,除去甲醇中的低沸点(或与甲醇沸点相近)的醛与酮杂质,精馏后的馏出物再用氢氧化钠作化学处理剂改善甲醇中的“酸度”指标。我们所采用的新工艺是用高锰酸钾和浓硫酸作化学催化剂,综合改善甲醇中的各项技术指标。这主要是利用高锰酸钾能氧化甲醇中的许多杂质的原理,有效地把低于或与甲醇沸点相近的醛与酮杂质氧化成沸点较高的羧酸,即把影响“酸度”技术指标的低沸点(或与甲醇沸点相近)的有机杂质氧化成高沸点杂质,以确保馏出物甲醇符合分析纯标准,同时改善甲醇的“易炭化物质”、“羰基化合物”、“还原高锰酸钾”等技术指标。
我们在做药用乙醇的挥发性杂质分析,遇到了问题,甲醇和乙醛的分离度不好,甲醇的峰也容易拖尾,将分流比调大是可以达到分离度的要求,但是又达不到检出限了,请大侠们帮帮忙,那些条件应该怎么设置
做乙醇挥发性杂质,甲醇与乙醛的峰分离不明显。求高手支招
甲醇,又称“木醇”或“木精”。是无色有酒精气味易挥发的液体。有毒,误饮5~10毫升能双目失明,大量饮用会导致死亡。 如果做实验时甲醇洒到手上或皮肤上会有什么危害呢?这个一直困扰,望大家给出意见!不胜感激
怎么分开甲醇和乙醛、氯乙烯色谱峰?我用SE-30(FID)分不开这几种物质?
最近在检测氨基乙醛缩二甲醇这个产品,用一个滴定的方法,颜色不好判断,哪位高手指点一下,最好有什么先进的方法,比如GC。

请问固定污染源排气中的甲醇和丙烯醛怎么做?对应国标是HJ/T33-1999和HJ/T36-1999。国标里都是填充柱做的,而且是进标气的。我用的仪器是岛津GC-2014,柱子是安捷伦的db-wax柱,有什么方法可以把甲醇和丙烯醛的标气融进一个溶液里,可以是同一种溶液也可以不同,目地就是做成液体来方便进样。有能容甲醇或是丙烯醛的溶液吗?[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2020/06/202006011738536376_7099_3862253_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2020/06/202006011738542003_1441_3862253_3.png[/img]
各位版友,我现在做新版药典乙醇的挥发性杂质准备工作,请问一下:1.配制对照品溶液的苯,乙醛,乙缩醛和4-二甲基-2-戊醇四种试剂需要什么级别的,多大纯度以上的可以达到要求?2.配制对照品用的无水甲醇可不可以用进口的色谱纯甲醇代替,如果可以是不是用色谱甲醇更好一些?
请问各位高效液相色谱流动相配制过程中甲醇是否可以用分析纯的代替。曾经比较过色谱醇和分析纯甲醇高效液相色谱图区别的最有发言权。如果经验丰富,请予以指导,甚为感谢!如果一般不行的解释是苍白的。谢谢各位!
做乙醇挥发性杂质,乙醛和甲醇峰形变得很奇怪,去年做的和现在做的差别很大,方法色谱柱一样,不知道哪里出问题了,谢谢各位老师。[img]https://ng1.17img.cn/bbsfiles/images/2020/10/202010261353589392_3447_5041548_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/10/202010261353589548_8430_5041548_3.png[/img]
为什么测试全氟调聚醇FTOH要先用9混(含全氟丙烯酸酯标线的标线初筛,再用FTOH标线定量[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2021/04/202104182031210404_7859_5195540_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2021/04/202104182031281487_5726_5195540_3.png[/img]
色谱纯甲醇的制备 随着液相色谱的广泛应用,作为反向液相色谱流动相的色谱纯甲醇的需求量与日俱增,产品供不应求。 但色谱纯甲醇要求纯度很高,且在紫外波段(220~280nm) 内需很高的透光率。 取200ml 工业甲醇,再加入1g2 ,4 - 二硝基苯肼(分析纯) ,回流25h ,过滤、精馏,取64. 5~65 ℃馏分 取200ml (工业级经上述实验纯化后) 的甲醇注入500ml 圆底烧瓶,加入11. 2g ,研磨后的分析纯KOH ,加2ml 蒸馏水,搅拌使之全溶,再加少许沸石,回流5h 精馏,取64. 5~65 ℃馏分,馏出液澄清。收集废液(实验2) 蒸干,用马沸炉于600 ℃灼烧2h ,除有机物,KOH 即可再生。 利用工业乙腈试制色谱纯乙腈 工业乙腈经氧化、精馏、吸附等化学物理方法能获得色谱纯乙腈,产品质量达标,性能可靠,能满足高效液相色谱分析的要求。 色谱纯乙腈的纯度要求达到99.9%’ 以上,有机物含量极低。工业乙腈中含有微量的氢氰酸、丙烯腈、乙醛、丙酮、丙腈、甲醇、丙烯醛、! (" )—甲基丙烯腈、顺式和反式丁烯腈、烯丙醇、水等杂质,需要采取化学物理方法进一步除去这些杂质,达到,-./ 级质量要求。 在1000ml 石英玻璃反应瓶中加入工业乙腈、氧化剂,在碱性条件下加热回流!1" 小时,将反应后的工业乙腈蒸馏,除去前、后馏分,再将获得的中间馏分进行吸、附或再蒸馏就可获得合格的色谱纯乙腈,前、后馏分可以返回到反应瓶中重复使用。
用FFAP分析白酒中乙酸乙酯和甲醇时,乙酸乙酯和甲醇的出峰时间比较接近,如果乙酸乙酯含量太高,甲醇含量太低的时候,乙酸乙酯出峰覆盖甲醇峰,无法对甲醇的浓度做出判断!请问该用什么方法解决?
用innowax柱子,7890 FID检测白兰地中甲醇,做得比较高,很担心柱子的分离效果,出峰顺序:甲醇前面有一个未知峰,甲醇峰,溶剂峰(乙醇)网上听很多人说,乙酸乙酯、乙缩醛和甲醇会分不开。跪求各位大侠还有什么方法判定它是否为甲醇。
请问专家:我用DB-1701,FID,载气:He 要用什么样的条件才能醋酸乙烯中的,乙醛,甲醇,乙醇分离开呢?
乙醇挥发性杂质检测,对照溶液b中的乙醛峰不明显,感觉和甲醇没有分开
做白酒时样品乙酸乙酯 乙缩醛 甲醇分不开。用的是innowax的柱子。仪器条件是:初温40保持1min,以4的速度升到130.以20的速度升到220保持1min







 我要推广仪器
我要推广仪器
 下载APP
下载APP