用邻苯二甲醛做氨基酸的衍生试剂,结果发现甲醇溶解的邻苯二甲醛比乙醇溶解的二甲醛衍生效果要好。请问谁能解释一下这个现象吗?
大家好,请问对二硝基苯和邻二硝基苯的纯物质可以溶于甲醇吗?请问有谁试过,主要是想配混标,其中间二硝基苯买的标液是甲醇中的标液。
为了测定邻氨基苯酚中微量杂质苯胺,现有下列固定相:硅胶,ODS键合相,流动相有。水-甲醇,异丙醚-己烷,应选用哪种固定相,流动相?为什么?
邻氨基苯甲酸甲酯液相检测我用甲醇:水+75:25,峰形不好看,拖尾,请老师给予指导调整一下流动相。谢谢!
大家谁知道用什么样的c18反相柱子能分开 邻硝基苯甲醇 和 邻硝基苯甲醛啊!!!!!!!
使用PEG-20M 30*0.32*0.25测定,汽化200,检测220,柱箱150,样品为邻氯对甲砜基甲苯(原料为四甲砜基甲苯)熔点90度,沸点280左右,溶剂为甲醇。测定谱图为3分钟溶剂出来,峰高700mv,12分钟样品出来,但是峰高只有1mv,我想请教下,是否柱子不合适,纯邻氯对甲砜基甲苯和甲醇溶剂体积比是1:1,为什么主峰那么低,是什么原因?非常感谢!
分离甲醇做溶剂中苯 甲苯 乙苯 二甲苯(邻间对)苯乙烯都有哪些色谱柱可以分开主要是甲醇中苯分不开,甲醇峰太大,影响苯的出峰,我不能换溶剂,只能用甲醇
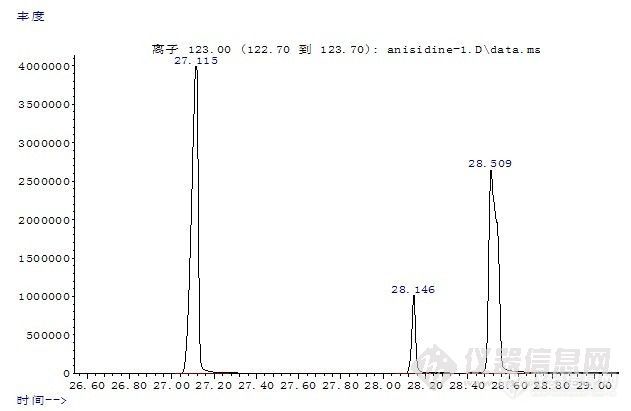
邻氨基苯甲醚同分异构体的分离摘要:为了实现邻氨基苯甲醚同分异构体的完全分离,作者采用气相色谱-质谱联用方法,通过改进色谱条件,使其取得了完全分离。试验表明采用气-质联用方法可以实现邻氨基苯甲醚同分异构体的完全分离。关键词:邻氨基苯甲醚;分离;异构体The Separation of O-Anisidine and Its isomersAbstract: In order to achieve the complete separation of o-anisidine and its isomers, the authors used gas chromatography - mass spectrometry methods, by improving the chromatographic conditions, so as to achieve their complete separation. The results showed that the gas-chromatography-mass spectrometry method can achieve the complete separationof o-anisidine and its isomers. Keywords: o-Anisidine; separation; isomers 1 前言采用气相色谱-质谱联用方法检测染料产品中23种有害芳香胺含量,按照现行国家标准GB/T17592-2006《纺织品禁用偶氮染料的测定》中提供的色谱条件,不但检测时间很长,而且大多数同分异构体保留时间相同或非常接近,同时这些异构体的质谱图又非常相似,从而造成无法使用特征离子对其进行定性和定量。如采用其它分析手段共同鉴别,如薄层色谱、液相色谱等,需要重新寻找条件,而且更换仪器费时费力,且多数实验室不一定同时具备这些设备。作者对芳香胺邻氨基苯甲醚及其异构体分离的问题进行了研究,其中,邻氨基苯甲醚属于有害芳香胺,而其同分异构体不属于有害芳香胺。作者通过改进色谱条件,使以上化合物达到很好的分离,提高了检测效率,减少了检测过程中的假阳性检出。2 试验2.1 仪器与试剂气相色谱-质谱联用仪(GC-MS):Agilent 7890A/5975C,美国Agilent公司毛细管柱:DB-17MS柱(30m×0.25mm×0.25μm)乙醚、硫酸亚铁等以上试剂均为分析纯;甲醇 色谱纯 美国Fisher公司旋转蒸发仪 上海亚荣生化仪器厂邻氨基苯甲醚及其同分异构体均为德国Dr.Ehrenstorfer公司。2.2 仪器操作条件色谱柱:DB一17MS 30m×0.25mm×0.25μm;温度:进样口220℃ ;辅助器280℃;离子源230℃ ;四极杆温度:150℃;柱温:40℃保持2分钟,以15℃/分钟升温至85℃ ,保持20分钟,再以30℃/分钟升至280℃,保持0分钟;载气:He;流速:1.0ml/分钟;离子化方式:EI;质量扫描范围:35—350;进样方式:不分流进样;进样0.2μL。2.3 执行标准试样处理按国标GB/T17592-2006执行。3 结果与讨论http://ng1.17img.cn/bbsfiles/images/2010/11/201011021932_256750_1604317_3.jpg图1 邻(间、对)氨基苯甲醚总离子流图由图1可以看出,邻氨基苯甲醚及其异构体在本实验条件下,可以得到很好的基线分离。保留时间分别为:邻氨基苯甲醚(27.115分钟);间氨基苯甲醚(28.509分钟);对氨基苯甲醚(28.146分钟)。在对染料产品中23种有害芳香胺含量的测定过程中,由于异构体的存在,并且多数异构体的质谱图很相似,如果保留时间相同,往往会形成假阳性结果的产生。当前针对有害芳香胺的气相色谱/质谱检测方法,大多采用非极性或极性较弱的色谱柱,如HP-5MS,DB-5MS,DB-35MS,这些色谱柱普遍存在的缺点是对常见的芳香胺异构体不能很好的分离。本方法通过使用中等极性色谱柱DB-17MS(固定相等同于50%苯甲基聚硅氧烷),同时使用三阶程序升温,很好的解决了这个问题。通过上面的数据与国标GB/T17592-
我做了邻氨基苯甲酸芳樟酯的谱图,可是里面只见到了芳樟醇和邻氨基苯甲酸甲酯,根本没有邻氨基苯甲酸芳樟酯的踪迹。我想请教一下,这是咋回事呢?是否邻氨基苯甲酸芳樟酯很难出峰?
请教各位前辈,鞋材纸板检测邻苯,用超声波提取,萃取剂为甲醇,提取效果如何?
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
高手们帮帮我吧。。苦恼了一个多月了。我用的安捷伦7820,做苯系物。只要测苯,甲苯,乙苯,邻间对二甲苯。。。用的甲醇做溶剂。热解析进样。用的强极性柱子DB-WAXETR 30米*0.32毫米*1.0微米条件是 进样口:150度,分流10:1(20:1和40:1都试过) 恒流模式:3.0mL/min 柱温:程序升温50度保持7分钟,10度每分钟升到110,保持5分钟 检测器:250度 尾吹30mL/min买的甲醇中苯系物的标准样品1mg/mL,配成的100微克/毫升,50,20,10,5,0……的浓度,进1微升结果除了甲醇和苯出峰超差,别的都分开,三个在一起的峰勉强分的还行吧。关键溶剂峰甲醇峰拖尾很厉害,在拖尾上就出苯峰了,而且苯峰特别小,50浓度的时候就已经很小了,再低浓度已经没法做了。。上个关于甲醇和苯的出峰图,高手们帮我看看吧。
我用的Agilent的7890GC,做的是甲醇中7种苯系物的含量测定,分别为苯、甲苯、乙苯、对二甲苯、间二甲苯、邻二甲苯、苯乙烯。柱子是用的瓦里安的52CB的毛细管柱,30m*0.32mm*0.45um,为了把对间二甲苯分开,设了个升温程序,50度保持4分钟,然后升温到80度保持至结束,载气流速2ml/min,分流比设的50。之前也试过别的条件,发现甲醇的溶剂峰非常大,苯的出峰总是在甲醇的溶剂峰的拖尾上,可能给定量带来较大误差,请问这个怎么解决?(换过手头的Waxetr柱,分对间二甲苯效果还不如这个柱,别的柱子更不行)
高手们帮帮我吧。。苦恼了一个多月了。我用的安捷伦7820,做苯系物。只要测苯,甲苯,乙苯,邻间对二甲苯。。。用的甲醇做溶剂。热解析进样。用的强极性柱子DB-WAXETR 30米*0.32毫米*1.0微米条件是 进样口:150度,分流10:1(20:1和40:1都试过) 恒流模式:3.0mL/min 柱温:程序升温50度保持7分钟,10度每分钟升到110,保持5分钟 检测器:250度 尾吹30mL/min买的甲醇中苯系物的标准样品1mg/mL,配成的100微克/毫升,50,20,10,5,0……的浓度,进1微升结果除了甲醇和苯出峰超差,别的都分开,三个在一起的峰勉强分的还行吧。关键溶剂峰甲醇峰拖尾很厉害,在拖尾上就出苯峰了,而且苯峰特别小,50浓度的时候就已经很小了,再低浓度已经没法做了。。上个关于甲醇和苯的出峰图,高手们帮我看看吧。
N-苯代邻氨基苯甲酸与N-苯基邻氨基苯甲酸这二种试剂是不是一样的。那位教师告诉我一下。谢谢!
邻苯混标,溶剂是二氯甲烷和正己烷,由于混标的溶剂容易挥发,稀释的时候我想用甲醇,可以吗?其实知道甲醇不合适,极性强但是又不知道该用什么稀释,求大神帮忙,谢谢了。
我目前是个实习生,要做毕业课题,是空气中苯系物的测定。现在是用甲醇做标样,甲醇中苯系物(332408)浓度为237ppm.做曲线是把甲醇稀释成5个浓度点做的。现在已经把苯。甲苯。乙苯。对间二甲苯。邻二甲苯。异丙苯的标准曲线做出来了。我做的标线浓度是10,30,50,70,90ug/ml。可是师傅要求我做方法检出限,我一窍不通,请大家帮帮忙,教我一下。最好说清楚整个过程
我们发的考核样是测定苯及其同系物 ,以甲醇为溶剂 ,甲醇拖尾太严重,苯的峰出不来 ,甲苯是一个骑峰,而且 对间二甲苯还分不开 ,邻二甲苯倒是没任何问题。 我调节了 载气流量 最小只有20ml/min 。柱温最低调到40°了,苯还是没出峰。 很多老师说 以二硫化碳为容积好一点 。但是省里发的考核样就是 甲醇作为溶剂啊 我也不知道该怎么办了。 我们FID只有填充柱的进样口。
2-氨基-4-氨基苯甲醚和2-硝基-4-氨基苯甲醚,这2个东西,我用非极性柱,换了几种不同型号的柱子,用甲醇,乙腈,四氢呋喃不同溶剂做流动相都分不开,我记得在氨基上可以上个什么保护基团,好像是十六烷基磺酸钠什么的,具体怎么搞忘了,请哪位高人指教...
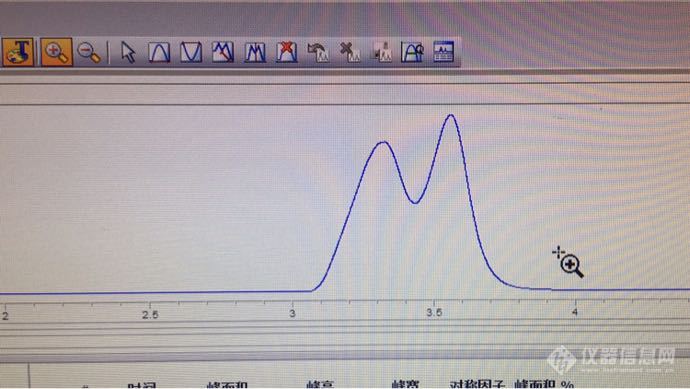
第一次做苯系物用的DB-624 分不开对 间二甲苯,苯乙烯和邻二甲苯,换了DB-WAX后其他都分离的不错 就苯出在甲醇的拖尾上,手头还有DB-1、DB-1701还有如图两个柱子 哪个大神做过好的分离 仪器是GC-7890B 顶空进样 [img=,690,388]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221410227003_2706_3086423_3.png[/img][img=,690,1225]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221410233313_5212_3086423_3.png[/img][img=,690,1225]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221410240663_942_3086423_3.png[/img]
我按gb17378.5《海洋监测规范:沉积物》中测有机碳的方法,配制苯基代邻氨基苯甲酸指示剂,将0.5克苯基代邻氨基苯甲酸溶于2g/L的碳酸钠溶液中。但是苯基代邻氨基苯甲酸只溶解了一点点,加热也不行。做过相关实验的同行,请指点一二。
苯基代邻氨基苯甲酸是什么颜色的,今天买来的苯基代邻氨基苯甲酸有的怪怪的
用7820A来做苯系物的标线,用的是甲醇中的9种苯系物标液,柱子是DB-WAX,60米,0.25mm的柱子,要分离甲醇跟苯的条件是什么?

前段时间买了瓶苯甲醇,主要是想检验,验证下样品中是否真含有苯甲醇,大概定量。就采购了一瓶苯甲醇,分析纯的,结果在质谱上发现买来的苯甲醇不是苯甲醇,是苯甲酸甲酯,我就傻了,如果我不是用气质分析,用气相,那误认为买的苯甲醇是正确的,那我的这个分析还敢拿出去见人吗?伤不起呀!http://simg.instrument.com.cn/bbs/images/brow/em09507.gif由于我们是代购的,对方还给我们说找他们了,就一个小厂,不承认,没开封是可以退,但开封了就不能退了,还有这种说法?代购又帮我们从国药那里拿了一瓶,还要我们重新买。心里好不爽,影响我们的时间和实验进度,更让我们不放心了。 http://simg.instrument.com.cn/bbs/images/brow/em09504.gifhttp://ng1.17img.cn/bbsfiles/images/2012/03/201203151445_354920_1608025_3.jpg315了,很想给他曝光,不过一瓶试剂也就55元的东西,不能坑人呀。当然理赔什么的还算小事,但怎么杜绝买的试剂让我们放心,好头痛。 http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif现在新购来了的国药的苯甲醇。
我做过一年的地表水中挥发酚,用的是4-氨基安替比林分光光度法,值得注意的是4-氨基安替比林容易变质,要用苯进行提纯。具体方法如下:将4-氨基安替比林置于干燥的烧杯中,加约10倍量的苯,用玻璃棒充分搅拌,并使块状物粉碎,将溶液连同沉淀移至干燥滤纸上过滤,再用少量苯洗至滤液为淡黄色为止。将滤纸上的沉淀物摊于表面皿,利用通风柜的机械通风,在较短的时间内使残留的苯挥发,去除后,把表面皿上的4-氨基安替比林用药瓶装好,置于干燥器内避光保存。(注意操作都要在通风柜中进行)[em01]
[em58] 本人做有机合成,通过2,4-二氨基苯甲醚的单酰化来合成2-氨基-4-乙酰氨基苯甲醚,但生成对位酰化物的同时,也形成邻位酰化物以及二酰化物,需要使用液相色谱方法进行分析,但尝试了甲醇,乙腈和水的不同配比流动相,始终无法将几个峰分开,上次根据一位大虾的建议尝试了乙腈与0.01M磷酸缓冲溶液的流动相,效果仍然不行,恳求各位大虾给与意见和方法,谢咯~~~~~~~!
各位大哥,小弟请教下你们邻氨基对甲苯酚怎么测定的啊·~~~是气相·色谱吗···沸点多少啊,用程序还是恒温啊·~·
0.2g N-苯基代邻氨基苯甲酸0.2g碳酸钠的100ml水中,加热溶解是先溶解0.2g碳酸钠,还是0.2g N-苯基代邻氨基苯甲酸,有顺序吗?再问一下,热水温度控制在多少?
最近在检测氨基乙醛缩二甲醇这个产品,用一个滴定的方法,颜色不好判断,哪位高手指点一下,最好有什么先进的方法,比如GC。
看到有一些测试塑料中有机锡总含量的方法,用甲醇作为萃取剂,但是做加入一定量的二乙基二硫代氨基甲酸钠(铜试剂),请问有什么用处?







 我要推广仪器
我要推广仪器
 下载APP
下载APP