关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
溴乙酸乙酯样品,用DMSO溶解,进样Agilent 7890 + DB624柱 + FID样品不出峰,何解?bp 溴乙酸乙酯159°,DMSO189°难道溶剂与样品发生了什么反应?
求文献:乙二胺与乙酸反应制备2-甲基咪唑啉
测定乙酸乙酯残留,溶剂为二甲基亚砜,测定精密度,发现有一些溶剂峰没有出现,而有的溶剂峰却出现了。请教大家是怎么回事,应该主溶剂的峰都是应该出现的啊,而且很大。溶剂峰和乙酸乙酯峰分离度很好。乙酸乙酯出峰在10分钟,二甲基亚砜出峰在17分钟。
大家好,我想用GC检测2甲基乙酸丁酯的左右旋异构体,试验了安捷伦的CycloSil-B手性柱,柱温40保持10分钟,1度/分升至80度。但结果左右旋异构体不能分开,仍然是一个峰。查了一些文献,都是二维气相,用串联的两根柱子来检测香精的左右旋异构体的,体系显得比较复杂。请问只用一根手性柱可以检测左右旋异构体吗?我应该怎么做呢?非常感谢。
哪位高人有甲苯、苯、甲醇、乙酸乙酯、N-甲基吡咯烷酮的检测标准,望分享,不胜感激!
请问DMBC Acetate乙酸二甲基苄基原醇酯的质谱图是怎样的?CAS000151-05-3
如题,在2.9min时乙酸乙酯出峰,在17min左右N,N-二甲基乙酰胺出峰,出峰时间为10min,峰形特别不好,四分之一圆形,我用的是岛津-2010,进样口和检测器温度均为200度,柱温为70度,分流比为10,急求!!!谢谢各位前辈!!
[color=#444444]请教一下谁用过GC-FID检测溴乙酸乙酯呀?是用的什么色谱柱呀?色谱条件是什么呀?有没有相关的文献介绍呀?谢谢各位大神!!![/color]
请问乙酸的甲基在氘代氯仿中的化学位移在多少啊?谢谢
请问乙酸的甲基在氘代氯仿中的化学位移在多少啊?谢谢
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=127755]对羟甲基苯氧乙酸的合成[/url]
各位大侠,本部门在开展自来水106项中二氯乙酸,三氯乙酸任务时,使用甲基叔丁基醚4mL萃取酸化后(pH2)40mL水样中的二氯乙酸,三氯乙酸,后取用其中2ml甲基叔丁基醚与2mL(10%硫酸酸化的)甲醇于45℃水浴中反应降温保护卤乙酸甲酯后,使用7mL150g/L的硫酸钠水溶液对其进行萃取问题来了,硫酸钠溶液萃取前,有机溶剂剩余4mL(甲醇2mL,甲基叔丁基醚2mL),萃取之后却发现有机相大大缩水,仅剩余1mL以下萃取的目的是为了除去有机相中的甲醇,保留甲基叔丁基醚,结果发现,大量甲基叔丁基醚消失,怀疑形成甲醇-甲基叔丁基醚-水共溶,因此,做了验证试验,加入2mL甲醇,2mL甲基叔丁基醚于分液漏斗,加入7mL150g/L硫酸钠溶液,摇晃后静置15min,期间不加热,不加酸,结果留下的有机相也仅为1mL。请教诸位达人,中是否可能形成甲醇-甲基叔丁基醚-水共溶,诸位在卤乙酸的gc检测预处理过程中采用何种方法??急求,在线等
我今天按照国标GB/T5750.10-2006中的9测定二氯乙酸,进行的是加标回收。因为是用甲基叔丁基醚做的介质,质谱分析时有很多峰,我无法确定哪一个是二氯乙酸甲酯,自带的质谱库内也没有。请问哪位能帮我找一下二氯乙酸甲酯的质谱分析参数,谢谢。
谁告诉我下 巯基乙酸异辛酯 二甲基二氯锡 用GC什么检测器?什么柱子分析啊?
异氰基乙酸乙酯和N,N-二甲基苯胺是否可以用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测?有没有方法条件呢?
我一向认为乙酸乙酯卡尔费休滴定用普通试剂就可以了,有个羰基不等于醛酮.RdH的应用手册上也说直接滴定就可以了。但是梅特勒和万通的人都说要用醛酮试剂,有道理吗?
各位老师,小弟最近在实验室中用气质(瓦里安3800)测香精样品,但是在分析乙酸乙酯时发现找不到对应峰。于是跑了一针乙酸乙酯标样(浓度30ppm,溶剂甲基叔丁基醚),但是还是找不到对应峰,请各个专家指点一二,看看问题究竟出在哪儿。谱库中乙酸乙酯的图谱及标品分析后的谱图如下,请参考。乙酸乙酯的标准质谱图:http://ng1.17img.cn/bbsfiles/images/2017/01/201701231132_01_2297325_3.bmphttp://ng1.17img.cn/bbsfiles/images/2017/01/201701231132_01_2297325_3.bmp乙酸乙酯标样图谱http://ng1.17img.cn/bbsfiles/images/2017/01/201701231134_01_2297325_3.bmp
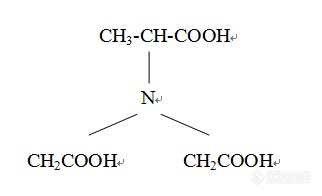
[color=#444444]甲基甘氨酸二乙酸(如下图所示)的检测方法,打算用液相色谱,[/color][color=#444444]但不知道溶剂该选啥,色谱柱怎么选择[/color][color=#444444]望高人指点[/color][color=#444444]不胜感激~[/color][color=#444444][img=,322,196]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021109562684_9156_1752329_3.png!w322x196.jpg[/img][/color]
我用的是据二甲基硅氧烷的柱子,30m*0.25mm*0.25um,溶剂是乙酸乙酯,溶质是苯,两者出峰时间太接近,分离效果很不好进样口:200度检测器:240度程序升温:35度恒温15min各位大大有什么好的方法呢?
三氟醋酸 Trifluoroacetic Acid 〔CF3COOH=114.02〕本品为无色发烟液体 有吸湿性 有强腐蚀性。在水乙醇丙酮或乙醚中易溶.三氟乙酸别名三氟醋酸,是一种重要的脂肪含氟中间体,由于含有三氟甲基的特殊结构,因此使其性质不同于其他醇类,可以参与多种有机合成反应,尤其用于合成含氟的医药、农药和染料等领域,国内外需求量越来越大,已成为含氟精细化学品的重要的中间体之一。三氟乙酸(醇、醛)主要用于新型农药、医药和染料等的生产,在材料、溶剂等领域也有较大的应用开发潜力。三氟乙酸主要用于合成多种含三氟甲基和杂环的除草剂,目前可以合成多种带有吡啶基、喹啉基的新型除草剂;作为极强的质子酸,它广泛用于芳香族化合物烷基化、酰基化、烯烃聚合等反应的催化剂;作为溶剂,三氟乙酸是氟化、硝化及卤代反应的优良溶剂,特别是其衍生物三氟乙酰基对羟基和氨基的优良保护作用,在氨基酸和多肽化合物合成方面有着非常重要的应用;三氟乙酸作为制备离子膜的原料和改性剂,可大幅提高烧碱工业电流效率,延长膜的使用寿命;三氟乙酸还可合成三氟乙醇、三氟乙醛和三氟乙酐。
(1)原理试样经处理后,在pH6左右的溶液中,镉离子与二硫腙形成配合物,并经乙酸丁酯萃取分离,导入原子吸收仪中,原子化以后,吸收228.8nm共振线,其吸收值与镉含量成正比,与标准系列比较定量。(2)试剂 氨水、混合酸、1g/L二硫腙-乙酸丁酯溶液(称取0.1g二硫腙,加10mL三氯甲烷溶解后,再加乙酸丁酯稀释至100rnL,临用时配制)、2mo1/L柠檬酸钠缓冲液(称取226.3g柠檬酸钠及48.46g柠檬酸,加水溶解,必要时加温助溶,冷却后加水稀释至500mL,临用前用1g/L二硫腙-乙酸丁酯溶液处理以降低空白值)、镉标准储备溶液和标准使用液的配制与碘化钾-4-甲基戊酮-2法中的相同。(3)仪器原子吸收分光光度计。(4)分析步骤①试样处理对于谷类要去除其中的杂物及尘土,必要时除去外壳。对于豆类,取可食部分洗净晾干,切碎充分混匀。②样品消化称取5.00g上述试样,置于250mL高型烧杯中,加15mL混合酸,盖上表面皿,放置过夜,再于电热板或电砂浴上加热。消化过程中,注意勿使干涸,必要时可加少量硝酸,直至溶液澄明无色或微带黄色。冷后加25mL水煮沸,除去残余的硝酸至产生大量白烟为止,如此处理两次,放冷。以25mL水分数次将烧杯内容物洗入125mL分液漏斗中。取与处理样品相同量的混合酸、硝酸按同一操作方法做试剂空白试验。③萃取分离 吸取0、0.25mL、0.50mL、1.50mL、2.50mL、3.50mL、5.0mL镉标准使用液(相当于0、0.05μg、0.1μg、0.3μg、0.5μg、0.7μg、1.0μg镉)。分别置于125mL分液漏斗中,各加盐酸(1+11)至25mL。向试样品处理溶液、试剂空白液及镉标准溶液各分液漏斗中各加5mL柠檬酸钠缓冲液(2mol/L),以氨水调节pH至5~6.4,然后各加水至50mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),以氨水调节pH至5~6.4,然后各加水至501mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),振摇2min,静置分层,弃去下层水相,将有机层放入具塞试管中,备用。④测定测定方法与碘化钾-4-甲基戊酮-2法中的相同。⑤结果计算 样品中镉的含量按下式进行计算。X=/(m×1000)式中,X为试样中镉的含量,mg/kg;A1为测定用试样液中镉的质量,μg;A2为试剂空白液中镉的质量,μg;m为试样质量或体积,g或mL。计算结果保留两位有效数字。⑥精密度 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%。
如何防止有机相中10mM乙酸铵的沉淀?我们流动相加了不少盐,但发现向纯乙腈中加10mM乙酸铵时不久会析出,如果用90%乙腈会影响洗脱。可以向乙腈中加二甲基亚砜吗?
二氯苯甲酸、乙酸等用什么色谱柱?样品中含二氯苯甲酸、乙酸、乙酸丁酯、醋酸铜、四甲基乙二胺、4-甲基吡啶应该怎么处理?用什么色谱柱?用安捷伦6890 FID色谱仪
用GB/T5750.10-2006的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]检测水中的二氯乙酸和三氯乙酸。萃取衍生过程和标准方法一样,用的是100ug/mL的混标,配制的标准曲线点两组分浓度一样。问题:1.为什么三氯乙酸的峰比二氯乙酸峰小呢?标准上三氯乙酸的峰比二氯乙酸的峰大很多,况且三氯乙酸多一个氯响应应该更大才对塞2.有做过这个实验的老师,麻烦问哈你们用的甲基叔丁基醚是哪个厂生产的啊,有没有卖色谱纯的厂家给推荐一个,还有内标1,2-DBP也是有没有色谱纯的?感觉我的基线比较毛燥。[img]https://ng1.17img.cn/bbsfiles/images/2022/07/202207081336599901_2592_2887366_3.png[/img]
RT最近接了个任务,让我用离子色谱测乙酸乙酯和苯肼中的氯、溴、硫酸根离子,浓度都比较低,问下怎么前处理。
4-氯乙酰乙酸乙酯的检测标准及检测方法N,N-二乙基乙二胺、N,N-二甲基乙二胺、N-乙基乙二胺系列产品,质量按Q/320801GHD001-2004标准执行、含量min.99.%
用作医药、农药中间体、生化试剂、有机合成试剂。三氟乙酸用于合成含氟化合物、杀虫剂和染料。是酯化反应和缩合反应的催化剂;羟基和氨基的保护剂,用于糖和多肽的合成。还用作选矿剂。用于有机合成。三氟乙酸是一种重要的脂肪含氟中间体,由于含有三氟甲基的特殊结构,因此使其性质不同于其他醇类,可以参与多种有机合成反应,尤其用于合成含氟的医药、农药和染料等领域,国内外需求量越来越大,已成为含氟精细化学品的重要的中间体之一。主要用于新型农药、医药和染料等的生产,在材料、溶剂等领域也有较大的应用开发潜力。三氟乙酸主要用于合成多种含三氟甲基和杂环的除草剂,可以合成多种带有吡啶基、喹啉基的新型除草剂;作为极强的质子酸,它广泛用于芳香族化合物烷基化、酰基化、烯烃聚合等反应的催化剂;作为溶剂,三氟乙酸是氟化、硝化及卤代反应的优良溶剂,特别是其衍生物三氟乙酰基对羟基和氨基的优良保护作用,在氨基酸和多肽化合物合成方面有着非常重要的应用,用于多肽合成中除去氨基酸的叔丁氧羰基(t-boc)保护基;三氟乙酸作为制备离子膜的原料和改性剂,可大幅提高烧碱工业电流效率,延长膜的使用寿命;三氟乙酸还可合成三氟乙醇、三氟乙醛和三氟乙酐。室温下三氟乙酸汞使氟苯起汞化反应(亲电取代),也可将腙转化为重氮化合物。此酸的铅盐可将芳烃转化为酚。可部分溶解二硫化碳和六碳以上烷烃,是蛋白质和聚酯的优良溶剂。它也是有机反应的优良溶剂,可获得在一般溶剂中难以获得的结果,例如喹啉在一般溶剂中催化氢化时,吡啶环优先氢化,但在三氟乙酸中苯环优先氢化。三氟乙酸在苯胺存在下分解成氟仿和二氧化碳。在HPLC中的应用:在反相色谱分离多肽和蛋白质的实验中,使用三氟乙酸 (TFA) 作为离子对试剂是常见的手段。流动相中的三氟乙酸通过与疏水键合相和残留的极性表面以多种模式相互作用,来改善峰形、克服峰展宽和拖尾问题。三氟乙酸与多肽上的正电荷及极性基团相结合以减少极性保留,并把多肽带回到疏水的反相表面。以同样的方式,三氟乙酸屏蔽了固定相上残留的极性表面。三氟乙酸的行为可以理解为它滞留在反相固定相的表面,同时与多肽及柱床作用。三氟乙酸优于其他离子修饰剂的原因是它容易挥发,可以方便地从制备样品中除去。另一方面,三氟乙酸的紫外最大吸收峰低于200nm ,对多肽在低波长处的检测干扰很小。改变三氟乙酸的浓度,可以细微地调整多肽在反相色谱上的选择性。这一影响对于优化分离条件、增大复杂色谱分析(如多肽的指纹图谱)的信息量是非常有益的。三氟乙酸添加在流动相中的浓度一般为 0.1% ,在这个浓度下,大部分的反相色谱柱都可以产生良好的峰形,当三氟乙酸浓度大大低于这个水平时,峰的展宽和拖尾就变得十分明显。三氟乙酸在分离蛋白等大分子的时候效果很好,在实际使用中,大家对于三氟乙酸的浓度都很难控制好,因为它是挥发性的物质,如果配置时间长了,就会挥发一些,改变了浓度。配制好以后一定要封闭好,防止挥发。
我根据国标在做二氯乙酸、三氯乙酸的检测项目,但是衍生出来的东西好像是有问题的,请各位看看我的衍生条件有没有什么问题:取25ml水,加入1.5ml浓硫酸,一勺硫酸钠,内标和标准,之后加入4ml左右的甲基叔丁基醚,震摇。取上清液3ml,再加入现配的衍生试剂(浓硫酸:甲醇为1:9)2.5ml。之后放在50℃水浴锅中恒温水浴2h,取出来4℃冷却。加入4ml碳酸氢钠,剧烈震摇,取上清液,上气相检测。请各位帮我看看有什么问题,做的时候需要什么注意事项!!!多谢
大家好,我急求用岛津气相色谱分离,乙酸和乙酸乙酯的分离条件,柱子,进样口,检测器温度,以及乙醇和乙酸乙酯的分离条件,柱子采用RTX-17ms,急求,谢谢大家!!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP