苯环上3位和5位上各带一个三氟甲基,扫碳谱后非常的杂,如何才能比较准确地找出相对应的峰并计算耦合常数呢?三氟甲基上的碳是否裂分位一个4重峰了,临位的碳也被裂分为双峰了呢?
求助给位,我想问一下四丁基溴化铵、十六烷基三甲基溴化铵、苄基三甲基溴化铵怎么做红外,它们都很容易潮解的,吸水性很强,我问一下,做红外的时候样品怎么处理,或者这些物质的标准红外数据在哪可以查到啊
三甲基氢醌与三甲基苯醌之间的关系,怎么计算两者之间的含量
[size=18px]目前在用AB的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测三苯基氯甲烷,Q1 MI模式扫243.1的离子[font=-apple-system, BlinkMacSystemFont, &](应该是三苯甲基碳正离子)[/font],发现基线非常高(30万-50万之间),且不稳定,时高时低,导致峰面积也 不稳定,打电话问客服,几个人几种说法,“液相部分污染了”“这个是正常现象,多走走就稳定了”,尝试用MRM模式去做,打出一个165.2的碎片,基线不到1000,做了线性和回收也都挺好,但是,这个碎片离子是怎么打出来的比较困惑,就怕以后再做的时候重现不出来……[/size][size=18px]流动相是90%甲醇,溶剂是正丁醇:乙腈(80:20)[/size][size=18px]请教一下各位大神,AB的仪器用SIM模式选择Q1 MI还是Q3 MI好呢?基线高且时高时低,除了污染还有什么原因呢?[font=-apple-system, BlinkMacSystemFont, &]三苯甲基碳正离子在质谱里能被打碎吗?会裂解成什么碎片离子?[/font][/size][size=18px][font=-apple-system, BlinkMacSystemFont, &][/font][/size]
请教各位大侠有没有做:臭氧危害物質(CFC/HCFC/Halon),氟溴烃,單甲基二氯二苯基甲烷,單甲基二溴二苯基甲烷 (DBBT),單甲基四氯二苯基甲烷 的检测方法,有的话能不能给我传一份,我的mail是wei.gao@isti.ocm.cn。万分感谢!
十六烷基三甲基溴化铵?请教大虾:十六烷基三甲基溴化铵 溶液需要在什么条件下保存?我配置8g/l 的溶液放置半个月左右后性质就不稳定了,溶液中主要成分有氯化钡,含量100g/l,我是在80摄氏度左右的热水中将十六烷基三甲基溴化铵和氯化钡溶解后定容常温保存。
三甲基溴化锍检测方法?是用离子色谱法还是液相?
十六烷基三甲基溴化铵含量及游离胺的测定方法.
十六烷基三甲基溴化铵是否溶于二氯甲烷,氯仿或者四氯化碳阿?在其他溶剂中的溶解度如何?谢谢了
液相色谱峰刚开始有正峰和反峰连在一起,乙腈和水做流动相的,是正常的吗,分析的2.4.6-三甲基苯甲酰氯含量甲醇酯化做的
我用滴定对苯醌的方法(溶液加碘化钾,盐酸,暗处静置后用硫代硫酸钠滴定)测三甲基苯醌含量,但是终点总是反色,找不到终点,请问高人们有解决的办法吗?谢谢!
[color=#444444]产品中残留有少量三溴苯酚钠,与空气氧化成红色;怎样才能检测出三溴苯酚钠的残留量呢?[/color][color=#444444]之前的一个思路:将产品用二氯甲烷溶解,加盐酸酸化,三溴苯酚钠转化成三溴苯酚,再用液相色谱分析;但是出峰很小,非正常产品含量0.06%左右,正常产品含量0.01%,但是后期再用相同的方法检测,三溴苯酚不出峰了,感觉很奇怪,请大神解答。。。[/color]
各位老师,哪位知道十六烷基三甲基溴化铵或季铵盐类物质的液相色谱分析条件,选用什么分析柱?哪个检测器好?请指导下呀。多谢了!
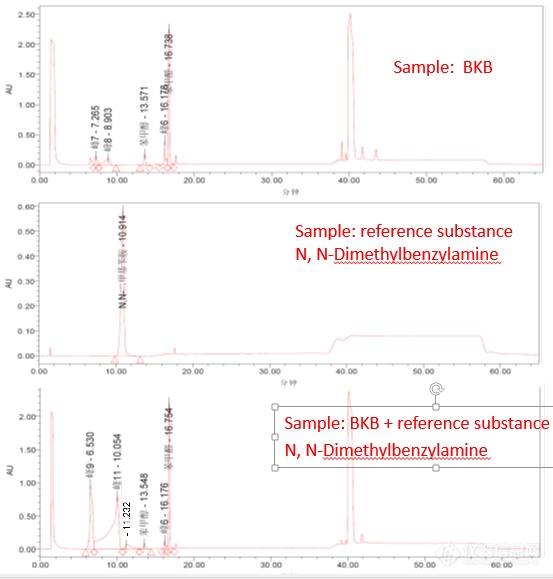
[color=#444444]最近在测苯扎溴胺(BKB)中苄基二甲基胺的残留,在测试条件下标样苄基二甲胺能够正常出峰,而进BKB样品时,苄基二甲基胺却不出峰,在BKB样品中加入苄基二甲基胺标样,却出现了两个奇怪的峰见色谱图,不知这是什么情况?[/color][color=#444444]色 谱 柱:端基封闭的C18(150×4.6mm,5μm)。[/color][color=#444444]柱 温:30℃[/color][color=#444444]流动相A:磷酸盐缓冲液(取己烷磺酸钠1.09g、磷酸二氢钠6.9g,溶于适量水中,用磷酸调节pH至3.5,用水稀释至1000ml,摇匀,即得。[/color][color=#444444]流动相B:甲醇[/color][color=#444444]时间(分) 流动相 A(V/V) 流动相 B(V/V)[/color][color=#444444]0-10 80 20[/color][color=#444444]10-14 80-50 20-50[/color][color=#444444]14-35 50 50[/color][color=#444444]35-36 50-20 50-80[/color][color=#444444]36-55 20 80[/color][color=#444444]流 量:1.0ml/min[/color][color=#444444]检 测 器:二极管阵列检测器(DAD)[/color][color=#444444][img=,553,579]https://ng1.17img.cn/bbsfiles/images/2019/06/201906031402122584_6731_1823055_3.png!w553x579.jpg[/img][/color]
有的书上说,三氟甲基在碳谱中可产生1:3:3:1的四重峰,距离比较远。本人做过一次,基本上只得到两个较高峰,两边的小峰有扫不出来的可能吗?而且,三氟甲基对alfa碳有裂分吗?
我做中控转化率,需要把二甲基亚砜与间羧基苯甲酸分离开,现正在寻找最佳的流动相比例.原先的比例是70+30+0.1+0.2(甲醇+水+醋酸+磷酸),但是分不开,出现包峰.望各位指导.
小弟做了好几次2-溴,3,3‘-二甲氧基联苯的氢谱,都发现甲基有三重峰,做何解?谢了!
我想了解4-氯-2-三氟甲基苯腈(4-Chloro-2-(trifluoromethyl)benzonitrile,CAS#320-41-2)的理化性质,但在网上只找到沸点109 º C (10 mmHg),是液体还是固体看不出来。因为这个沸点是真空条件下的。那位老师有相关的信息,请告诉我,谢谢!
我们分析2.4.6-三甲基苯甲酰氯,我的岛津液相出峰开始会出个倒峰是什么原因,紫外 C18 乙腈水 1ml/min
请问有朋友遇到类似的问题吗;岛津的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]前两天做曲线时低浓度的两个点1PPM,2PPM的2,4,6三溴苯酚(替代物)峰形出现不正常,后面从4PPM开始又恢复正常了。今天做标准曲线时,2,4,6三溴苯酚(替代物)在12PPM 这个浓度点开始332、330离子出来的峰图形就开始不正常了。 15PPM这个浓度点332、330离子的峰图形就完全不正常了。出现了锯齿状的一堆峰。后面接着走的空白样品,样品,加标样品也是这一替代物的峰形不正常。这种问题怎么解决?[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2024/03/202403071117162971_8517_6098339_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2024/03/202403071117325182_9341_6098339_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2024/03/202403071117326728_6036_6098339_3.png!w690x387.jpg[/img]
1,1,2-三甲基环丙烷与HBr加成后,生成的产物是2,2-二甲基-3-溴丁烷?
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
谁有丁二酸酐,三乙胺,对甲基苯磺酸的检测方法
使用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]检验甲基苯丙胺,突然峰出的很差,低浓度直接没有峰了,其他东西正常。
5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。
[color=#444444]高效液相色谱法测定十六烷基三甲基溴化铵的环境参数是多少?[/color][color=#444444]例如柱温,甲醇浓度,最大吸收波长[/color]
[color=#444444]高效液相色谱法测定十六烷基三甲基溴化铵的环境参数是多少?[/color][color=#444444]例如柱温,甲醇浓度,最大吸收波长[/color]
GB2763中关于亚砜磷的限值要求,标准中指出残留物为亚砜磷 砜吸磷 甲基内吸磷之和,以亚砜磷表示,不知道亚砜磷 砜吸磷 甲基内吸磷这三者之间有什么关系,是可以相互转化,还是同分异构体?
水质中的三溴苯酚一共有几种?







 我要推广仪器
我要推广仪器
 下载APP
下载APP