谁有丁二酸酐,三乙胺,对甲基苯磺酸的检测方法
聚山梨酯80残留溶剂检测中的(三乙胺与二甲基甲酰胺),三乙胺的出峰面积越来越大。不知道大家有没有碰到过这种情况,出峰面积一针比一针大,求指点!
测二甲基,乙醇,三乙胺用什么柱子比较好?
顶空[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]做残留溶剂三乙胺的回收率为什么会降低?具体如下:第一针进溶剂5毫升二甲基亚砜,第二针进标液(500mg三乙胺用二甲基亚砜定容100毫升,取5毫升用二甲基亚砜定容到59毫升),第三针称0.5g样品加二甲基亚砜5毫升,第四针称0.5g样品加5毫升第二针的标液。
三乙胺我们在生产中用作缚酸剂,现在得到一个三乙胺邻二氯苯混合溶液,配置了不同浓度的三乙胺溶液,但是打出来同样溶度的三乙胺峰面积相差挺大的,这个是为什么啊?
做溶剂残留,用的月旭的wm624色谱柱,混合对照其他峰都有,就三乙胺没有峰,溶剂是nn二甲基甲酰胺,顶空进样
顶空[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测三乙胺,谁有相关经验吗?设置条件大概是多少?三乙胺溶液里面需要添加碱溶液吗?求回复
有什么比较好的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]方法将六甲基二硅氧烷(硅醚)和三乙胺分开
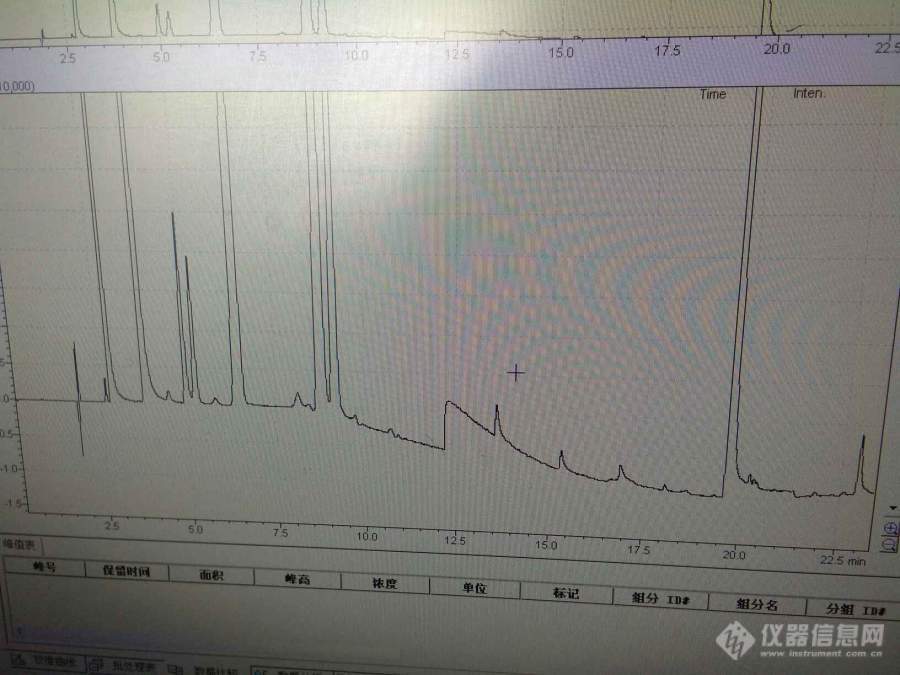
[color=#444444]我想请问一下,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中,顶空进样,溶剂为N-甲基吡咯烷酮,柱子DB-624,溶液里面成分有甲醇,乙醇,乙腈,二氯甲烷,正己烷,乙酸乙酯,四氢呋喃,三乙胺,吡啶,DMF,目前三乙胺峰判断不出来,也不确定到底有没有出来峰,图谱已传上,请教一下[/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/06/201906141009051727_9546_1752329_3.jpg!w690x517.jpg[/img][/color]
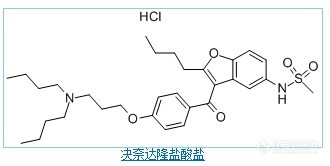
盐酸决奈达隆 结构式我做这个样品的时候存在严重的拖尾现象1、乙腈:水=55:45 峰分布出来,大概需要10分钟左右才能出一个趴着的峰2、乙腈:水(0.02mol/L 磷酸二氢钠)=55:45 拖尾3.33、乙腈:水(0.02mol/L磷酸二氢钠):三乙胺=550:450:1 拖尾2.1 此时PH=7.284、乙腈:水(0.04mol/L磷酸二氢钠):三乙胺=550:450:1 拖尾2.1 此时PH=7.12我觉得这两个基团(甲基磺酰胺 另外一个氨基 )会和ODS填料的硅羟基形成氢键,所以加入了三乙胺扫尾,效果是稍微好了一点,但是拖尾因子到2.1时候不再改善,我先加入磷酸将PH调整到2.5,后又用三乙胺将PH调整到7,峰型又有所改善,感觉是因为加大了三乙胺的量,可是网上查的三乙胺最大添加量为1ml/L,也没有见到有先将PH调低再加入大量三乙胺的做法求教各位大大,这种基团应该怎么处理,有什么思路吗,在线等,万分感谢http://ng1.17img.cn/bbsfiles/images/2011/04/201104072058_287643_1638724_3.jpg
各位老师。我最近在做一个化合的异构体分离,流动相中需要加如三乙胺扫尾,但是发现采用三乙胺扫尾后,色谱峰比不加三乙胺的保留时间要延迟很多,这是什么原因呢?我一直认为三乙胺是不会影响手性保留的!大家有什么高见?
最近在做水质三乙胺的检测,发现校准曲线很难做直,请问主要有哪些影响因素,还有三乙胺的标样何处可以买到?
刚接触分析没多长时间,公司领导要求我做一个车间回收三乙胺定量的方法,面积百分比不准,然后就尝试做一个外标法,因为试样当中有邻二氯苯和三乙胺两种物质,所以我就配置了不同浓度的三乙胺溶液,5%.10%.15%.20%.25%.的溶液,用自动进样器进样,发现峰面积相差很大,这是为什么?到底该怎么做呢?求助前辈~我的气谱方法是这样的,进样0.2ul,分流比100:1,进样口气化温度250,检测器300,柱箱150保持5分钟。HP-5中性柱,安捷伦7820A,FID检测器
要测一个原料药中三乙胺的残留。仪器是安捷伦的6890N,7694E,条件如下:色谱柱HP-1,柱温100,进样口250,分流比1:1,柱压7.7psiFID250顶空平衡30min,温度70/100/110三乙胺浓度16ug/ml(用水溶),取2ml至10ml顶空瓶我的问题是我进了三乙胺后跟了一针空白,三乙胺的位置有出峰,再跟一针空白就没有了,我试着做进样精密度,连续进了三针峰面积从29涨到了40,RSD非常差。而且换了DB-624的柱子,这个问题还是存在,哪位大侠指点一下!
小弟分析生物碱,用的流动相为甲醇/水(含0.14%H3PO4和0.5%三乙胺),防拖尾效果不错。 但0.5%三乙胺是不是太高了?会对柱子不好吗?望论坛高手释疑
有这么一种流动相,甲醇:水:三乙胺:乙酸=600:400:1:1那么加三乙胺和乙酸的作用是什么?
请问:二甲基疏基乙胺国内有否厂家生产此产品?能否提供相关的分析此产品的信息?
[color=#444444]三乙胺-丙酮-盐的混合液,三乙胺的含量在5%以下,请问有什么好的方法可以测定三乙胺的含量,在实验室用氢火焰毛细柱打可以出峰,但是如果含量特别低时担心[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]打不出,导致微量三乙胺检测不出来。查文献有溴酚蓝分光光度法、红外色谱,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]、电位返滴定法,以及混合液放入盐酸,用过量NaOH滴定未反应酸,溴酚蓝做指示剂等方法。请问这几种方法哪个比较准确,并且好操作,谢谢各位了![/color]
有这个标准:取样品0.5g,精密加浓氨试液-甲醇(1:20),冷浸1小时,加热回流1小时。按这个方法处理出来的样品峰形很难看,而且他离度也不好。它所用的流动相是甲醇:0.1%磷酸溶液(用三乙胺调PH至6.0)。同样是含碱性,为什么处理样品不用三乙胺-甲醇(1:20)?
三乙胺怎么除水呢,我要求不高,查资料说不能用硫酸镁,可以用氯化钙吗
在液相分析中,有时会用到三乙胺的,看到资料说三乙胺对色谱柱的影响是不可逆的,但有些说是好好冲洗对柱子影响不是很大的,到底怎么样呢?大家用的话,一般的加入量是多少呢,用完后又是怎么冲洗的呢?
用三乙胺做扫尾剂,一般它的量加多少呢?
是否有脂肪族胺类分析方法?是否有三甲胺,三乙胺,三丙胺,正丁胺标气购买?[em0805]
利用顶空-[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测试三乙胺含量,感觉前后两针互相干扰,进完一针样品后,再进空白,会有三乙胺的峰,然后再进一次空白三乙胺的峰面积会减小,应该是三乙胺残留了,请问有什么办法让三乙胺不残留。
我现在测生物碱(吗啡)的含量,分离度可以了但老是拖尾,三乙胺的量都加到0.1%了,还能不能再增加了?再增加有效果吗?三乙胺肯定能消除拖尾现象吗?各位有做过生物碱的吗?传授一下经验咯。我实在是着急呀,各位大侠帮帮忙呀,盼望中…………
在用液相色谱分析某些酸性药品时,有的时候会在流动相中加入三氟乙酸,加入三氟乙酸的目的是什么啊?有时候还会在流动相中加入二乙胺,目的是什么啊?仅仅是要调节pH值么?还有就是,二乙胺可以用三乙胺代替么?哪位高手给解答一下啊,具体点的。
请问有没有人测过三乙胺的微量水分?我看到有人用容量法可以测,直接就拿卡尔费休试剂滴定,也能出结果。不知道胺类的产品会不会和卡氏试剂起反应哦?
谁有三乙胺的GB标准吗?
哪个厂家有三乙胺(色谱纯)我急需求购!!!!!!!
通常液相色谱分析中,在流动相中加乙酸和三乙胺,大家来谈谈,在什么情况下需要加乙酸,什么情况下加三乙胺,加多少是怎么控制的?







 我要推广仪器
我要推广仪器
 下载APP
下载APP