我公司现生产的一种产品,四甲基哌啶醇是精制而成包装时需对进行挥发份进行测定,但该产品易于升华,故现在还没有一个合适的方法,测定溶剂是否已烘于。
三丙酮胺沸点205四甲基哌啶胺沸点188-189[img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037318102_4160_3963991_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037319030_2268_3963991_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037317834_4061_3963991_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037318341_2757_3963991_3.png[/img]
有没有人检测过N-甲基哌啶
2010年版药典氟哌啶醇片红外鉴别方法是:取本品粉末适量(相当氟哌啶醇约50mg),用20ml三氯甲烷分次研磨使溶解,过滤,取滤液水浴蒸干,残渣减压干燥后,压片,与标准图谱比较. 因为氟哌啶醇易溶于三氯甲烷,我在做的过程中就没有剥片;另氟哌啶醇对光 热具不稳定性,滤液采用60度水浴蒸至近干,采用药典原料项下的方法干燥:即60度减压干燥,约5小时,但取出压片时发现它的性状为半固体,压出的片透明度不好,还粘模具,没法做! 会不会是辅料有影响呢?想请教一下,如何改进我的试验,压制一个好的片子?
哌啶和乙醇
春节好 求助氮氧自由基哌啶醇含量化验方法 谢谢!
本人要测定一样品中的哌啶含量,以哌啶纯品进样时出峰,以DMF做纯品的溶剂配成1mg/ml溶液时,哌啶不出峰,DMSO作溶剂时情况一样,但在大约1ml的DMF中加入两三滴纯哌啶时,哌啶出峰,而且峰面积不少,大概换算到1mg/ml的浓度也应该可以检出(1滴大概4~5mg),请问为什么会这样呢?大家有检过哌啶吗,用的是什么溶剂呢?我的检测条件是DB-624,进样口200度,分流比10:1,恒流2ml/min,程序升温40度(5min),15°C/min,220(5min),检测器FID250度,载气是氮气,请大家指教下!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
请问有哪位高人测过聚四亚甲基醚二醇中羰基比的。能给个红外谱图吗?
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
本人在岛津GC-2014上做样,FID检测器, 进样口:200,检测器:250,此样品以乙醇为溶剂,溶解以样品限度为浓度的吡啶0.004mg/ml 、哌啶0.004mg/ml、,在DB-624上做过,柱长:30m*0.32mm*1.80um, 进样:1.0ul 以40°C,8min,以20°每分钟升到200°C,保持3分钟,分流20:1、 10:1、5:1都测试过了,可哌啶就是不出峰,从浓溶液定位的图谱来看,哌啶和吡啶出峰时间相差0.1min,其实也就是难以分离,为此换hp-5柱子定位,可是哌啶和吡啶的峰总是拖尾,出峰时间虽然相差0.4min,但是实际按照以上的分流条件做样时候,哌啶不出峰,若是再修改分流比恐怕乙醇过载,杂质变大,更不好判断,于是又换了DM-1色谱柱,同样条件,浓溶液定位,这次哌啶在这柱子上根本不出峰,大浓度也不出峰,无奈又换了DM-WAX柱子做样,同样条件,同样定位,吡啶倒是峰形很好,可哌啶,居然引得基线上升,然后又持平,像是斜坡的样子,但是根本不积分!所以,本人想请问,各位兄台,各位高手,有没有一根柱子是对哌啶响应值高,且完美的分离吡啶和哌啶的,请指教!
2,4,7,9-四甲基-5-癸炔-4,7-二醇 化学不懂 不知道里面的数字代表什么意思?有盆友知道吗?
4-羟基哌啶用Aglient1260检测,流动相乙腈,甲醇,各种比例都试过,C18的长柱,短柱都用过,怎么都是不出主峰,求助给些线索!
我现在在合成双哌啶醇酯,反应之后无法分离形成的单酯和双酯,想通过滴定来判断单双酯的含量,请问各位大侠有没有什么好的方法!即测定六元环上的仲氨.
4-羟基哌啶溶于甲醇后,只有甲醇峰,样品再浓都没有?跟柱子有关系吗?
Hi: 有没有版友知道工业用聚四亚甲基醚二醇(PTMEG)GB/T 25254中羟基测定中配置乙酰化混合物的时候醋酸酐作为酰化剂,吡啶作为溶剂,稳定剂和缚酸剂,其中加的冰醋酸起什么作用,一定要加吗?
有哪位朋友做过哌啶基哌啶的分析,能否跟我联系下,有一些问题想请教谢谢
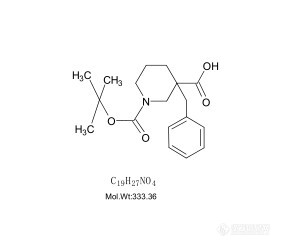
色谱世界的各位大侠们,1-boc-3-苄基哌啶甲酸文献报道说用4.6mm*150mm C18的二氧化硅柱子,用乙腈、水、三氟乙酸作为流动相,在214和254处有吸收峰。我们用紫外全波长扫描后,发现只有在204处有紫外吸收峰,可是我们用乙腈和水作流动相,这个东西在214处不出峰。这个东西该怎么检测纯度呢。这个化合物是通过1-boc-3-哌啶甲酸 和溴苄合成的, 还有1-boc-3-哌啶甲酸 的熔点是159-162℃。 我们这个东西的熔点是109-116℃。 所以这两个东西很定有是不一样的。 实在不行只有打核磁了。我们这个原料的紫外吸收也在204这个位置。但我们用254的紫外薄层检测,发现原料不显色,1-boc-3-苄基哌啶甲酸 轻微显色。[img=,281,247]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231115067664_5937_1815404_3.jpg!w281x247.jpg[/img]
1-BOC-4-哌啶甲酸检测其纯度时DB-624和HP-5严重拖尾,在DB-WAX中无法出峰,请问选择什么色谱柱合适?或有其它的检测方法
现在要做哌啶,N-甲酰基哌啶的含量,都是微量的,用了DB-624,DB-waxetr的测1000ppm的峰形都不好,请问有推荐色谱柱和方法么
有没有谁知道中国食品接触材料中对于改性PCT材料,需要做的2,2,4,4四甲基1,3环丁二醇的检测方法是什么
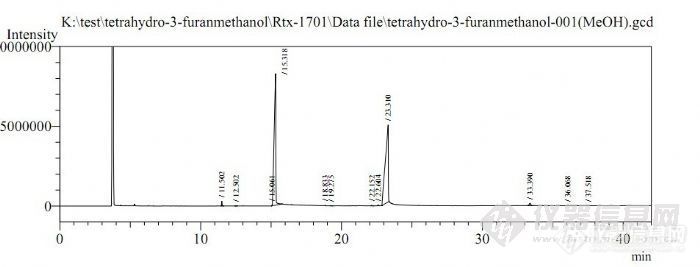
大家好,我在检测3-羟甲基四氢呋喃纯度的时候,因为对这个东西一点也不了解,也没有查到相应的资料,就先用了RTX-1701柱子检测,进样口:250,检测器(FID):300,分流比:20:1,线速度进样:1.0mL/min,升温程序:50(2)-10-250(20)),用甲醇稀释了一下,发现走得不怎么好,具体谱图如下,大家帮我看看,应该怎么做?知道的TX请不吝赐教,给我一个详细的分析方法,最好帮我也附张图谱!谢谢!比较着急!!!http://ng1.17img.cn/bbsfiles/images/2011/07/201107030846_302772_1973612_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP