甲基环戊二烯三羰基锰(MMT)气相色谱法检测方法本标准规定了甲基环戊二烯三羰基锰的分类、要求、试验方法、检验规则、标志、包装、运输、贮 存和安全。本标准适用于用作汽油抗爆剂的甲基环戊二烯三羰基锰。 分子式:C9H7MnO3 相对分子质量:218.09(根据2007年国际相对原子质量) 甲基环戊二烯三羰基锰含量的测定:在选定的工作条件下,样品经气化通过毛细管色谱柱,使其中各组分得到分离,用氢火焰离子化检 测器检测,用面积归一化法或内标法计算甲基环戊二烯三羰基锰的含量。 试剂:二乙二醇二甲醚。 无水乙醇。氢气:体积分数不低于 99.99%。 空气:经活性炭和分子筛净化。氦气:体积分数不低于 99.999%。仪器设备 :GC5890气相色谱仪,配氢火焰离子化检测器(FID),灵敏度和稳定性符合 GB/T9722 中的有关规定, 可进行毛细管色谱分析。N2000色谱工作站。色谱仪器型号GC5890型色谱仪 配有FID检测器毛细管色谱柱HP-5 30*0.32*0.25专用毛细管柱色谱工作站N2000 (电脑1台自备)气体装置氮氢空发生器 HGT300E1台或高纯氮、氢气、空气钢瓶各一瓶分析天平:感量 0.0001g。 5.8.3.4 进样器:5μL [font=
最近要测羰基指数,样品为PTMEG(聚四亚甲基醚二醇)是是四氢呋喃的聚合物。测它的羰基指数怎么弄?一般找那个峰为基准?现在只知道有文献上写羰基指数CI=Ac=o/Aref..... 求高人指点

[color=#DC143C]五羰基铁[/color][img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911232034_186121_1610969_3.jpg[/img]Fe(CO)5为CO与Fe的合成物,化学反应方程为:5CO+Fe→Fe(CO)5。 [color=#00008B]Fe(CO)5的物理性质:[/color] 1、铁属于过渡元素,在它的原子中产生充填不满结构的电子层,在与一氧化碳相互作用下形成Fe(CO)5时,由铁原子与5个CO分子组成中获取不足的电子。其分子结构式如图: 2、在常压下,Fe(CO)5的熔点在-20.3℃左右,沸点在103.6℃左右,临界温度286℃左右。在100℃以下没有明显分解,100℃-130℃约有1%的分解140℃ -160℃有3.3%弱分解,160℃特别是179以上时,普遍强烈分解。 [color=#DC143C] Fe(CO)5的化学性质:[/color] 1、Fe(CO)5完全溶解于汽油、苯、四氯化萘、苯醛、丙酮、溴化苯、二氯化苯和其它溶液。 2、从-15℃起火花时,羰基物蒸汽与空气混合物一定产生燃烧,在温度34℃ 时(亦有报道60℃)就在适当条件下能够自燃。 3、Fe(CO)5相当的活泼,容易形成氢化羰基物H2Fe(CO)4及其金属盐Na2Fe(CO)4,卤化羰基物Fe(CO)4I2、亚硝酰基羰基物Fe(CO)2(NO)2、氯化羰基物Fe(CO)3(CH3OH)、环戊二烯羰基物[C5H5Fe(CO)2]2等很多化合物。 4、Fe(CO)5光化学性能很好,在光的作用下Fe(CO)5分解形成Fe2(CO)9。 5、当加热到140℃时,Fe(CO)5易氧化,形成Fe2O3(铁氧体)。 6、针对Fe(CO)5的临界温度,在常压及温度在250℃-300℃时进行Fe(CO)5的热解,是Fe(CO)5最重要的应用,是工业化制取羰基铁粉的最基本方法。
样气里面含有硫化氢和羰基硫,但标气里面只有羰基硫,怎么标定色谱啊?
羰基加氢产物在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中的出峰位置是在原料峰前还是峰后。。目前做的是二苯甲酮催化剂加氢。。就是想做羰基加氢生成醇。。具体还是不是很清楚产物位置
羰基镍 Nickel carbonyl CAS:13463-39-3[color=#ff0000]理化性质[/color]具有霉味的无色至淡黄色易挥发液体。分子式C4-Ni-O4。化学式 Ni(CO)4。分子量 170.73。相对密度 1.318(17℃)。熔点 -19.3℃。沸点 43℃。闪点 -20℃。自燃点 93.33℃。蒸气密度 5.95(50℃)。蒸气压 53.32kPa(400mmHg 25.8℃)。蒸气与空气混合物可燃下限 2% 。水中溶解度为0.018g/100ml 不溶于稀酸、稀碱 溶于乙醇、苯、氯仿、丙酮、四氯化碳、王水、乙醚、硝酸。液态羰基镍侵蚀某些塑料、橡胶、涂层。空气中氧化,与氧化剂反应生成一氧化碳和相应的盐。遇热、明火、氧化剂易燃。20℃时,它的蒸气在空气和氧气中的分压达到2.00 kPa(15 mmHg)时爆炸 液态羰基镍在60℃时爆炸。不能与硝酸、氯、溴、可燃性蒸气共存。[color=#ff0000]消防措施[/color][color=#0000ff][size=5][sup] [/sup][/size][/color]消防人员须穿戴全身防护服。用雾状水、泡沫、二氧化碳、干粉灭火。[color=#ff0000]储运须知[/color]包装标志:毒害品。包装方法:(I)类。高强度玻璃瓶充一氧化碳或其他不反应气体,气密封口,装在金属罐内,周围以惰性吸收材料衬填,外木箱或钢瓶装。储存条件:储存于阴凉、干燥、通风良好的仓库内。远离热源和火源。避光储存。仓库温度控制在28℃以下。搬运时轻装轻卸,防止容器破损。[color=#ff0000]泄漏处理[/color]切断一切火源,戴好防毒面具等全部防护用品。用不燃性分散剂制成的乳液刷洗。如无分散剂可用砂土吸收,倒至空旷地方掩埋。对污染地面用肥皂或洗涤剂刷洗,经稀释的污水放入废水系统。[color=#ff0000]接触机会[/color]羰基镍主要用于精炼镍、制造丙烯酸和甲基丙烯酸酯、有机合成的催化剂、作为钢和其他金属涂层、在冶金和电子工业中用于汽相扩散渗镀。当一氧化碳通入金属镍可形成不稳定的羰基镍。[color=#ff0000]侵入途径[/color]]主要经呼吸道吸入,也能经皮肤吸收。[color=#ff0000]毒理学简介[/color]人吸入TCLo: 7 mg/m3 LCLo: 30 ppm/30M。大鼠吸入LC50: 35 ppm/30M(244mg/m3)。小鼠吸入LC50: 67 mg/m3/30M。羰基镍为高毒物质。兔吸入浓度为 291mg/m^3 后 5分钟,发现镍在肺、血和肾的滞留量分别为 38.1 %、11.5%及 7.9%,肝内含量甚微。三天内约可随尿排出吸收镍的 62.2%。给大鼠 LD50 的剂量,经静脉、皮下、腹腔投毒后,24小时内脏器官肉眼检查无变化,第二天可见肺、肝脏肿大。肺部病变表现为肺水肿和灶性出血,肺血管周围有炎症细胞侵润,肺泡上皮细胞肥大和增生,肺泡壁增厚。肝脏为肝小叶中央中度淤血。中枢神经系统水肿,大脑半球毛细血管出血。约经二周后存活动物病理变化可趋好转。有人曾对一例接触羰基镍后13天急性中毒死亡的管道装配工人进行尸检。肺主要病变为肺实质由于成纤维细胞侵润而使很多区域硬变,只有很少量含有空气的软区。
请问,含有羰基的物质,且极性较大,在反相色谱柱上几乎不保留,是否可以考虑用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析?常用的羰基[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]衍生化方法有哪些?注意事项有哪些?谢谢大家的帮助!
有没有版友用2,4-二硝基苯肼比色法做过油脂中的羰基值,测定结果精密度如何?需要注意哪些环节,我在测定过程中发现稳定性不好,不知什么原因
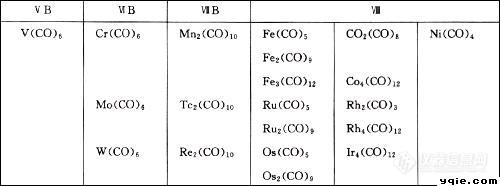
曾经在工作中接触过一点金属羰化物的红外分析,当时为了做好这项工作,我做了不少案头工作,现在我要离开分析行业了,整理了一下自己曾经做过的一些东西,真的还是有点留恋,就拿这些内容整理一下,作为原创大赛的最后一篇告别文。我这人其实挺烦为发表而作的八股文的,所以更喜欢比较自由的论坛帖子。好了,下面言归正传:一.基础知识 金属羰基化合物是指羰基和金属原子形成含σ-π配键的配位化合物,几乎全部的过渡金属可以和一氧化碳形成稳定的羰化物。这种配合物中金属离子的氧化态一般很低,有的甚至等于零,如4]、Na[Co(CO)[sub]4]、[Mn(CO)[sub]5Br]、Co[sub]2(CO)[sub]8]等。有些金属羰化物及其衍生物在一些有机合成中用作催化剂,有的已用于工业生产中,例如工业上常在高温高压下由合成气(CO+H[sub]2)与金属铑、钴或它们的盐合成羰化物作为催化剂。这里是别人列出的一些金属羰化物.[img]http://ng1.17img.cn/bbsfiles/images/2010/12/201012142027_267014_1640192_3.jpg[/img] 我接触到的是合成中用于均相催化剂的钴\铑\铱的羰基化合物.[/size]
哪位大神,知道产品:N,N'-羰基二咪唑 CDI CAS No- 530-62-1 质量检测方法?特别是下面两个指标的检测方法:1、Active Carbonyl Content 活性碳基的含量2、Free Imidazole Content自由咪唑基的含量
[color=#444444]ESI 质谱条件下,M+H的二级产生含羰基的加氢峰,m/z 220,M+Na的二级碎片产生含羟基的加钠峰m/z 244,二者相差24。羰基氧和羟基氧与钠离子和氢离子的结合能力是怎样?[/color][color=#444444]难道钠离子更容易稳定含羟基离子?氢离子更容易稳定含羰基的离子?该怎么解释呢?[/color]
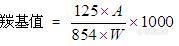
[size=3]前面已经对羰基值发过帖子,没怎么解决,再发一次。2010版药典一部对羰基值的计算公式作了重大修改:[/size][font=宋体][size=3][img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005252017_220628_1604723_3.jpg[/img]虽说对羰基值的计算公式作了修改,但具体到每味药材下,其限定值却没作修改。比如桃仁,其羰基值限定为11.请注意:设测得吸光度A=0.1(已经够小了),W=0.5g(附录中规定0.025-0.5g),最后计算得30.不合理。[/size][/font]
根据我所看到的谱图,乙酸的羰基碳会随着酸性增大向低场移动,而二苯甲酮的羰基碳会随着酸性增大向高场移动,这是为什么呢?
化学键合固定相的基本理论将有机官能团通过化学反应共价键合到硅胶表面的游离羟基上而形成的固定相称为化学键合相。这类固定相的突出特点是耐溶剂冲洗,并且可以通过改变键合相有机官能团的类型来改变分离的选择性。 1.键合相的性质目前,化学键合相广泛采用微粒多孔硅胶为基体,用烷烃二甲基氯硅烷或烷氧基硅烷与硅胶表面的游离硅醇基反应,形成Si-O-Si-C键形的单分子膜而制得。硅胶表面的硅醇基密度约为5个/nm2,由于空间位阻效应(不可能将较大的有机官能团键合到全部硅醇基上)和其它因素的影响,使得大约有40~50%的硅醇基未反应。残余的硅醇基对键合相的性能有很大影响,特别是对非极性键合相,它可以减小键合相表面的疏水性,对极性溶质(特别是碱性化合物)产生次级化学吸附,从而使保留机制复杂化(使溶质在两相间的平衡速度减慢,降低了键合相填料的稳定性。结果使碱性组分的峰形拖尾)。为尽量减少残余硅醇基,一般在键合反应后,要用三甲基氯硅烷(TMCS)等进行钝化处理,称封端(或称封尾、封顶,end-capping),以提高键合相的稳定性。另一方面,也有些ODS填料是不封尾的,以使其与水系流动相有更好的"湿润"性能。由于不同生产厂家所用的硅胶、硅烷化试剂和反应条件不同,因此具有相同键合基团的键合相,其表面有机官能团的键合量往往差别很大,使其产品性能有很大的不同。键合相的键合量常用含碳量(C%)来表示,也可以用覆盖度来表示。所谓覆盖度是指参与反应的硅醇基数目占硅胶表面硅醇基总数的比例。pH值对以硅胶为基质的键合相的稳定性有很大的影响,一般来说,硅胶键合相应在pH=2~8的介质中使用。 2.键合相的种类化学键合相按键合官能团的极性分为极性和非极性键合相两种。常用的极性键合相主要有氰基(-CN)、氨基(-NH2)和二醇基(DIOL)键合相。极性键合相常用作正相色谱,混合物在极性键合相上的分离主要是基于极性键合基团与溶质分子间的氢键作用,极性强的组分保留值较大。极性键合相有时也可作反相色谱的固定相。常用的非极性键合相主要有各种烷基(C1~C18)和苯基、苯甲基等,以C18应用最广。非极性键合相的烷基链长对样品容量、溶质的保留值和分离选择性都有影响,一般来说,样品容量随烷基链长增加而增大,且长链烷基可使溶质的保留值增大,并常常可改善分离的选择性;但短链烷基键合相具有较高的覆盖度,分离极性化合物时可得到对称性较好的色谱峰。苯基键合相与短链烷基键合相的性质相似。另外C18柱稳定性较高,这是由于长的烷基链保护了硅胶基质的缘故,但C18基团空间体积较大,使有效孔径变小,分离大分子化合物时柱效较低。 3.固定相的选择分离中等极性和极性较强的化合物可选择极性键合相。氰基键合相对双键异构体或含双键数不等的环状化合物的分离有较好的选择性。氨基键合相具有较强的氢键结合能力,对某些多官能团化合物如甾体、强心甙等有较好的分离能力;氨基键合相上的氨基能与糖类分子中的羟基产生选择性相互作用,故被广泛用于糖类的分析,但它不能用于分离羰基化合物,如甾酮、还原糖等,因为它们之间会发生反应生成Schiff 碱。二醇基键合相适用于分离有机酸、甾体和蛋白质。分离非极性和极性较弱的化合物可选择非极性键合相。利用特殊的反相色谱技术,例如反相离子抑制技术和反相离子对色谱法等,非极性键合相也可用于分离离子型或可离子化的化合物。ODS(octadecyl silane)是应用最为广泛的非极性键合相,它对各种类型的化合物都有很强的适应能力。短链烷基键合相能用于极性化合物的分离,而苯基键合相适用于分离芳香化合物。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=152826]大气中羰基化合物GC/MS分析方法[/url]摘要:介绍了一种灵敏度高、可靠并且能同时检测大气中2O种羰基化合物(C 一C 。)的分析方法.该方法是采用涂布PFPH(衍生剂)的TenaxTA作为固体吸附剂采集大气样品,然后再经过溶剂洗脱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/质谱(GC/MS)分离检测的一项分析技术.校正曲线的可决系数(R。)、检测限(LOD)、平行样标准偏差(RSD,n=6)、回收率分别为0.995—1.00,0.15—1.04ngm~ ,7.3% 一15.8% 和92.7% ~109.2%.该方法成功地应用到对大气中羰基化合物的定量检测.对羰基化合物浓度的日变化分析表明,上海大气中羰基化合物浓度变化与大气光化学反应的强弱有密切关系.
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=48984]ASTM E411-00 二硝基苯肼测定微量的羰基化合物.doc[/url]
检测煤气中的羰基硫,硫化氢,二硫化碳含量,但是采样时,发现气体中含有一定的水蒸气。用什么干燥剂能除去水蒸气,并且不于需要检测的三种气体反应。现在查到[color=#cc0000]无水氯化钙[/color]不与硫化氢反应,是否于另外两种气体反应没有查看。其他干燥剂也可以,主要是不于三种气体反应。 最好能反复使用。 TRANSLATE with x English [align=left] [table][tr][td][url=#ar]Arabic[/url][/td][td][url=#he]Hebrew[/url][/td][td][url=#pl]Polish[/url][/td][/tr][tr][td][url=#bg]Bulgarian[/url][/td][td][url=#hi]Hindi[/url][/td][td][url=#pt]Portuguese[/url][/td][/tr][tr][td][url=#ca]Catalan[/url][/td][td][url=#mww]Hmong Daw[/url][/td][td][url=#ro]Romanian[/url][/td][/tr][tr][td][url=#zh-CHS]Chinese Simplified[/url][/td][td][url=#hu]Hungarian[/url][/td][td][url=#ru]Russian[/url][/td][/tr][tr][td][url=#zh-CHT]Chinese Traditional[/url][/td][td][url=#id]Indonesian[/url][/td][td][url=#sk]Slovak[/url][/td][/tr][tr][td][url=#cs]Czech[/url][/td][td][url=#it]Italian[/url][/td][td][url=#sl]Slovenian[/url][/td][/tr][tr][td][url=#da]Danish[/url][/td][td][url=#ja]Japanese[/url][/td][td][url=#es]Spanish[/url][/td][/tr][tr][td][url=#nl]Dutch[/url][/td][td][url=#tlh]Klingon[/url][/td][td][url=#sv]Swedish[/url][/td][/tr][tr][td][url=#en]English[/url][/td][td][url=#ko]Korean[/url][/td][td][url=#th]Thai[/url][/td][/tr][tr][td][url=#et]Estonian[/url][/td][td][url=#lv]Latvian[/url][/td][td][url=#tr]Turkish[/url][/td][/tr][tr][td][url=#fi]Finnish[/url][/td][td][url=#lt]Lithuanian[/url][/td][td][url=#uk]Ukrainian[/url][/td][/tr][tr][td][url=#fr]French[/url][/td][td][url=#ms]Malay[/url][/td][td][url=#ur]Urdu[/url][/td][/tr][tr][td][url=#de]German[/url][/td][td][url=#mt]Maltese[/url][/td][td][url=#vi]Vietnamese[/url][/td][/tr][tr][td][url=#el]Greek[/url][/td][td][url=#no]Norwegian[/url][/td][td][url=#cy]Welsh[/url][/td][/tr][tr][td][url=#ht]Haitian Creole[/url][/td][td][url=#fa]Persian[/url][/td][td] [/td][/tr][/table] [/align] TRANSLATE with COPY THE URL BELOW [url=javascript:Microsoft.Translator.FloaterOnShareBackClick()]Back[/url] EMBED THE SNIPPET BELOW IN YOUR SITEEnable collaborative features and customize widget: [url=http://www.bing.com/widget/translator]Bing Webmaster Portal[/url] [url=javascript:Microsoft.Translator.FloaterOnEmbedBackClick()]Back[/url]
硫磺装置酸性气体含硫化氢、二氧化硫、氨气、羰基硫,二氧化碳等,我们分析用的GDX502柱子,外标法,TCD,用标气标定是二氧化硫和氨气分不开,求助,大家有经验的给予指导,谱图和分析条件设置?借鉴谢谢
[color=blue][b]化学键合固定相的基本理论[/b][/color]将有机官能团通过化学反应共价键合到硅胶表面的游离羟基上而形成的固定相称为化学键合相。这类固定相的突出特点是耐溶剂冲洗,并且可以通过改变键合相有机官能团的类型来改变分离的选择性。 [b]1.键合相的性质[/b] 目前,化学键合相广泛采用微粒多孔硅胶为基体,用烷烃二甲基氯硅烷或烷氧基硅烷与硅胶表面的游离硅醇基反应,形成Si-O-Si-C键形的单分子膜而制得。硅胶表面的硅醇基密度约为5个/nm2,由于空间位阻效应(不可能将较大的有机官能团键合到全部硅醇基上)和其它因素的影响,使得大约有40~50%的硅醇基未反应。 残余的硅醇基对键合相的性能有很大影响,特别是对非极性键合相,它可以减小键合相表面的疏水性,对极性溶质(特别是碱性化合物)产生次级化学吸附,从而使保留机制复杂化(使溶质在两相间的平衡速度减慢,降低了键合相填料的稳定性。结果使碱性组分的峰形拖尾)。为尽量减少残余硅醇基,一般在键合反应后,要用三甲基氯硅烷(TMCS)等进行钝化处理,称封端(或称封尾、封顶,end-capping),以提高键合相的稳定性。另一方面,也有些ODS填料是不封尾的,以使其与水系流动相有更好的"湿润"性能。 由于不同生产厂家所用的硅胶、硅烷化试剂和反应条件不同,因此具有相同键合基团的键合相,其表面有机官能团的键合量往往差别很大,使其产品性能有很大的不同。键合相的键合量常用含碳量(C%)来表示,也可以用覆盖度来表示。所谓覆盖度是指参与反应的硅醇基数目占硅胶表面硅醇基总数的比例。 pH值对以硅胶为基质的键合相的稳定性有很大的影响,一般来说,硅胶键合相应在pH=2~8的介质中使用。 [b]2.键合相的种类[/b] 化学键合相按键合官能团的极性分为极性和非极性键合相两种。 常用的极性键合相主要有氰基(-CN)、氨基(-NH2)和二醇基(DIOL)键合相。极性键合相常用作正相色谱,混合物在极性键合相上的分离主要是基于极性键合基团与溶质分子间的氢键作用,极性强的组分保留值较大。极性键合相有时也可作反相色谱的固定相。 常用的非极性键合相主要有各种烷基(C1~C18)和苯基、苯甲基等,以C18应用最广。非极性键合相的烷基链长对样品容量、溶质的保留值和分离选择性都有影响,一般来说,样品容量随烷基链长增加而增大,且长链烷基可使溶质的保留值增大,并常常可改善分离的选择性;但短链烷基键合相具有较高的覆盖度,分离极性化合物时可得到对称性较好的色谱峰。苯基键合相与短链烷基键合相的性质相似。 另外C18柱稳定性较高,这是由于长的烷基链保护了硅胶基质的缘故,但C18基团空间体积较大,使有效孔径变小,分离大分子化合物时柱效较低。 [b]3.固定相的选择[/b] 分离中等极性和极性较强的化合物可选择极性键合相。氰基键合相对双键异构体或含双键数不等的环状化合物的分离有较好的选择性。氨基键合相具有较强的氢键结合能力,对某些多官能团化合物如甾体、强心甙等有较好的分离能力;氨基键合相上的氨基能与糖类分子中的羟基产生选择性相互作用,故被广泛用于糖类的分析,但它不能用于分离羰基化合物,如甾酮、还原糖等,因为它们之间会发生反应生成Schiff 碱。二醇基键合相适用于分离有机酸、甾体和蛋白质。 分离非极性和极性较弱的化合物可选择非极性键合相。利用特殊的反相色谱技术,例如反相离子抑制技术和反相离子对色谱法等,非极性键合相也可用于分离离子型或可离子化的化合物。ODS(octadecyl silane)是应用最为广泛的非极性键合相,它对各种类型的化合物都有很强的适应能力。短链烷基键合相能用于极性化合物的分离,而苯基键合相适用于分离芳香化合物。 另外,美国药典对色谱法规定较严,它规定了柱的长度,填料的种类和粒度,填料分类也较详细,这样使色谱图易于重现;而中国药典仅规定填料种类,未规定柱的长度和粒度,这使检验人员难于重现实验,在某些情况下还浪费时间和试剂。来源:色谱网[em61]
将有机官能团通过化学反应共价键合到硅胶表面的游离羟基上而形成的固定相称为化学键合相。这类固定相的突出特点是耐溶剂冲洗,并且可以通过改变键合相有机官能团的类型来改变分离的选择性。 1.键合相的性质目前,化学键合相广泛采用微粒多孔硅胶为基体,用烷烃二甲基氯硅烷或烷氧基硅烷与硅胶表面的游离硅醇基反应,形成Si-O-Si-C键形的单分子膜而制得。硅胶表面的硅醇基密度约为5个/nm2,由于空间位阻效应(不可能将较大的有机官能团键合到全部硅醇基上)和其它因素的影响,使得大约有40~50%的硅醇基未反应。残余的硅醇基对键合相的性能有很大影响,特别是对非极性键合相,它可以减小键合相表面的疏水性,对极性溶质(特别是碱性化合物)产生次级化学吸附,从而使保留机制复杂化(使溶质在两相间的平衡速度减慢,降低了键合相填料的稳定性。结果使碱性组分的峰形拖尾)。为尽量减少残余硅醇基,一般在键合反应后,要用三甲基氯硅烷(TMCS)等进行钝化处理,称封端(或称封尾、封顶,end-capping),以提高键合相的稳定性。另一方面,也有些ODS填料是不封尾的,以使其与水系流动相有更好的"湿润"性能。由于不同生产厂家所用的硅胶、硅烷化试剂和反应条件不同,因此具有相同键合基团的键合相,其表面有机官能团的键合量往往差别很大,使其产品性能有很大的不同。键合相的键合量常用含碳量(C%)来表示,也可以用覆盖度来表示。所谓覆盖度是指参与反应的硅醇基数目占硅胶表面硅醇基总数的比例。pH值对以硅胶为基质的键合相的稳定性有很大的影响,一般来说,硅胶键合相应在pH=2~8的介质中使用。 2.键合相的种类化学键合相按键合官能团的极性分为极性和非极性键合相两种。常用的极性键合相主要有氰基(-CN)、氨基(-NH2)和二醇基(DIOL)键合相。极性键合相常用作正相色谱,混合物在极性键合相上的分离主要是基于极性键合基团与溶质分子间的氢键作用,极性强的组分保留值较大。极性键合相有时也可作反相色谱的固定相。常用的非极性键合相主要有各种烷基(C1~C18)和苯基、苯甲基等,以C18应用最广。非极性键合相的烷基链长对样品容量、溶质的保留值和分离选择性都有影响,一般来说,样品容量随烷基链长增加而增大,且长链烷基可使溶质的保留值增大,并常常可改善分离的选择性;但短链烷基键合相具有较高的覆盖度,分离极性化合物时可得到对称性较好的色谱峰。苯基键合相与短链烷基键合相的性质相似。另外C18柱稳定性较高,这是由于长的烷基链保护了硅胶基质的缘故,但C18基团空间体积较大,使有效孔径变小,分离大分子化合物时柱效较低。 3.固定相的选择分离中等极性和极性较强的化合物可选择极性键合相。氰基键合相对双键异构体或含双键数不等的环状化合物的分离有较好的选择性。氨基键合相具有较强的氢键结合能力,对某些多官能团化合物如甾体、强心甙等有较好的分离能力;氨基键合相上的氨基能与糖类分子中的羟基产生选择性相互作用,故被广泛用于糖类的分析,但它不能用于分离羰基化合物,如甾酮、还原糖等,因为它们之间会发生反应生成Schiff 碱。二醇基键合相适用于分离有机酸、甾体和蛋白质。分离非极性和极性较弱的化合物可选择非极性键合相。利用特殊的反相色谱技术,例如反相离子抑制技术和反相离子对色谱法等,非极性键合相也可用于分离离子型或可离子化的化合物。ODS(octadecyl silane)是应用最为广泛的非极性键合相,它对各种类型的化合物都有很强的适应能力。短链烷基键合相能用于极性化合物的分离,而苯基键合相适用于分离芳香化合物。另外,美国药典对色谱法规定较严,它规定了柱的长度,填料的种类和粒度,填料分类也较详细,这样使色谱图易于重现;而中国药典仅规定填料种类,未规定柱的长度和粒度,这使检验人员难于重现实验,在某些情况下还浪费时间和试剂。这就需要我们对色谱条件进行优化。
麻烦大家告诉一下测定羰基价时,苯精制后真空蒸馏所使用的温度?谢谢!!!:)
大家做羰基价用的苯都是重蒸的吗?是否可以直接 用色谱纯的呢??因为我们很少自己蒸馏试剂,没有专用的成套的玻璃装置,使用平时的蒸馏装置恐怕会有二次污染有这方面经验的版友给点建议啊!!
在测定桃仁羰基值,酸值时,用脂肪油提取时加多少正己烷合适?
2017年能力验证计划 CNAS PT0031-1603固体化工品 化学品熔点的测定 CNASPT0031-1702甲醇羰基化合物的测定羰基化合物02450247GB/T 6324.5报名截止日期:2017年6月31日,具体实施时间:2017.7-2017.11
好像醛酮和非醛酮的区分是以是否有羰基为条件的,叫羰基试剂岂不是更好?
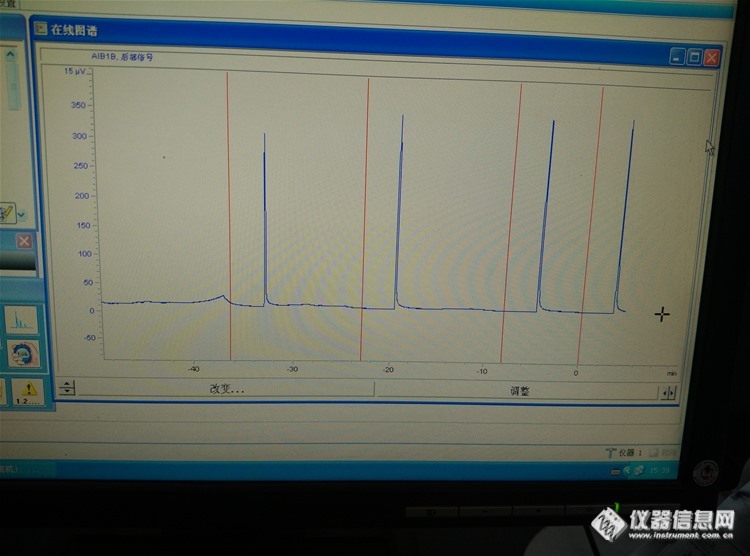
安捷伦7890SCD检测器。。问题是,羰基硫出峰,硫化氢不出峰,10ppm的。高含量的出峰,已经进过很多高含量的硫化氢了,10ppm的还是不出峰,500ppm的硫化氢出峰偏低,求了解SCD大神解救http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_667385_2114910_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016040716340464_01_2114910_3.jpg
药典附录中关于酸败度测定法,共有三个值需要测定:1、酸值;2、羰基值;3、过氧化值。此三个值均是在油脂的提取后,进行测定。这里主要讨论第二个羰基值的测定。关于羰基值的测定,需要用到有毒溶剂苯。为方便,以下为药典原文:羰基值的测定 羰基值系指每1kg供试品中所含羰基化合物的毫摩尔数。除另有规定外,取供试品0.025~0.5g,精密称定,置25ml量瓶中,加苯使溶解,稀释至刻度,摇匀。精密量取5ml,置25ml具塞试管中,精密加4.3%三氯醋酸的苯溶液3ml及0.05%二硝基苯肼的苯溶液5ml,混匀,置60度水浴中加热30分钟,冷却后沿管壁慢慢精密加入4%氢氧化钾的乙醇溶液10ml,密塞,剧烈振摇1分钟,放置10分钟,以相应试剂为空白,照紫外-可见分光光度法在453nm的波长处测定吸光度,照公式计算。因为05年版与10年版的计算公式相差太大,此处不录。我的问题是:假如苯中含有杂质,这杂质为小分子的含羰基化合物,这就影响到了测定。如果这些含羰基化合物是微量的,则可能不会影响测定。但我们在实验中,发现,即使不加供试品,相应试剂的颜色已经成了一种黑色了,致使光无法透过比色皿,而呈现以下的现象:在相当大的(大于5)吸光度范围内,光谱呈剧烈、快速频率的波动。我猜是因为光透不过比色皿而引起的。我想问:按照紫外-可见分光光度法下对溶剂的要求,此处测定波长为453nm,位于可见光区,以空气为空白,测定苯的吸光度,完全合格。但是,如果杂质的羰基化合物,这同样是测不出来的,也就是即使有杂质的羰基化合物,以453nm为检验溶剂是否合格,当然也就合格了。因为羰基化合物本身就没有颜色。所以,我觉得,此处应有其它规定,以检验苯是否真的合格。
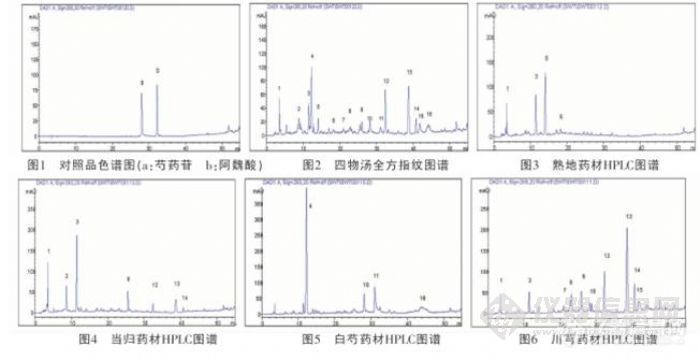
【作者】:雷艳青,雷 鹏,刘 韶,李新中【摘要】: 目的:为探讨中药配方颗粒在中药复方中应用的可行性,本研究选取补血名方“四物汤”为实验对象,对该方的传统饮片汤剂与配方颗粒汤剂进行中药指纹图谱的比较。方法:采用高效液相色谱、二极管阵列检测器:色谱柱为C18(Dia-monsil 250 mm×4.6 mm,5 μL);流动相中,流动相组分A为水,B为甲醇,C为0.05%磷酸;梯度洗脱,条件为组分C保持10%不变,组分B从0到90%;检测波长为280 nm;流速为1.0 mL·min-1;柱温:35 ℃。结果:同一批药材的两种汤剂其HPLC指纹图谱相似性好,相似度0.924。6批不同厂家的汤剂间指纹图谱基本一致,相似度在0.70-0.98。结论:方剂四物汤的中药配方颗粒与传统饮片汤剂比较,指纹图谱相似性好,提示内在化学成分基本相同。【作者单位】:湖南省脑科医院,中南大学湘雅医院【关键词】: 四物汤;传统饮片汤剂;配方颗粒汤剂;HPLC指纹图谱http://ng1.17img.cn/bbsfiles/images/2012/07/201207310921_380753_1838299_3.jpg

GB/T 5009.37-2003 食用植物油卫生标准 , 采用 2,4-二硝基苯肼比色法测定羰基价提供的计算公式中提到的各种醛的毫克当量吸光系数是什么意思?毫克当量是什么意思?http://ng1.17img.cn/bbsfiles/images/2016/07/201607082356_599865_2055165_3.png
各位高手请问怎样测羰基价准确啊?我今天测了一个样品,前后相隔不到十分钟,吸光度差好多,这可如何是好啊?还有样品吸光度居然比空白还低,我用的是吸光度相同的比色皿。







 我要推广仪器
我要推广仪器
 下载APP
下载APP