很多杂质不会出峰,但是对色谱柱有损害,你如何选择和考虑的呢?
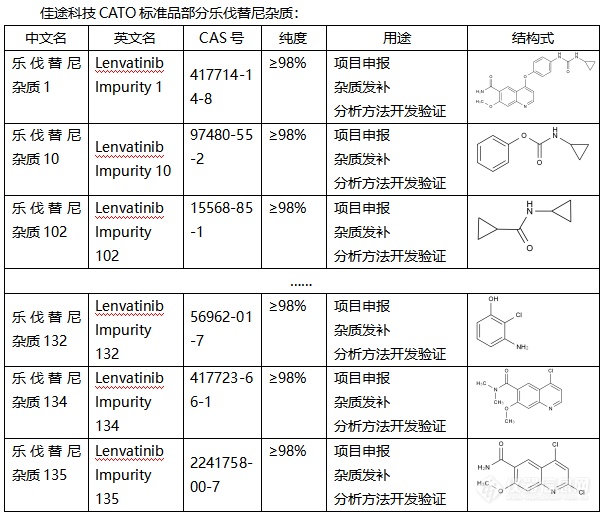
乐伐替尼,作为现代医学的瑰宝,广泛应用于肿瘤治疗领域。然而,就像其他药物一样,乐伐替尼在生产过程中也可能会产生杂质。这些微小的杂质,虽然量少,却可能对药物的疗效和安全性产生不可忽视的影响。为了确保乐伐替尼的纯净与安全,科学家们引入了CATO标准品进行杂质分析。CATO标准品,就像一把精准的尺子,能够帮助研究人员准确地检测和衡量乐伐替尼中的杂质。通过对比和分析,我们可以清楚地了解杂质的种类、数量以及可能对药物产生的影响。这项应用研究不仅提升了乐伐替尼的生产质量,更为患者的安全用药提供了有力保障。借助CATO标准品,我们能够及时发现并控制杂质,确保每一颗乐伐替尼都是纯净、有效的。未来,随着科学技术的不断进步,我们期待看到更多关于乐伐替尼杂质分析的研究成果,为患者带来更加安全、可靠的治疗方案。同时,也期待CATO标准品在更多药物杂质分析中发挥重要作用,守护人类的健康与安全。[img=,603,515]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021836237186_8744_6381568_3.png!w603x515.jpg[/img]广州佳途科技股份有限公司是一家专业的CATO标准品生产厂家,我们目前库存有全套乐伐替尼杂质,能够提供相应的系列图谱和产品COA证书,并支持溯源。我们公司已经通过了国内外双ISO 17034质量体系认证,欢迎广大客户选购。

在甲基培尼皮质醇的制造过程中,可能会生成一些杂质。这些杂质可能会出现在原料中,也可能在制药过程中的化学反应中产生。不论其来源,杂质的存在都可能影响到药物的质量、安全性和疗效。例如,甲基培尼皮质杂质可能增加药物的毒性,或导致不良反应。同样,杂质也可能对甲基培尼皮质醇的药效产生影响。因此,制药公司必须在生产过程中严格检测和控制这些杂质。检测和控制药品中的杂质是药品质量控制的重要组成部分。CATO标准品对杂质的研究不仅有助于保证药品的质量和安全性,也可以为优化制药过程提供参考。比如,通过对杂质的研究,可以找到产生这些杂质的原因,从而改进制药过程,减少杂质的生成。[img=,600,588]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052058222428_1748_6381668_3.png!w600x588.jpg[/img]
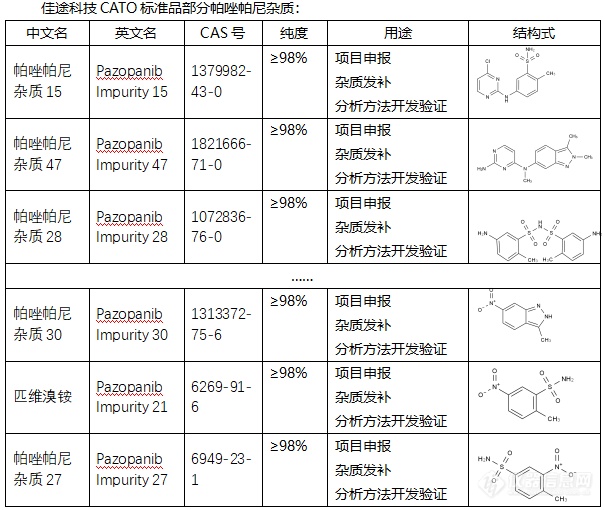
◇帕唑帕尼杂质 帕唑帕尼杂质是在帕唑帕尼药物制备或存储过程中可能产生的物质。帕唑帕尼杂质有多种,其中一些具有特定的CAS号、化学式和分子量。例如,帕唑帕尼杂质(Pazopanib Impurities)的CAS号为59816-94-3,化学式为C22H22N8,分子量为398.46。此外,帕唑帕尼杂质还包括一些异构体和其他结构类似物,如Pazopanib Isomer等。 CATO标准品提供的帕唑帕尼全套的杂质,这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分。[img=,605,510]https://ng1.17img.cn/bbsfiles/images/2024/02/202402192050040241_6306_6381607_3.png!w605x510.jpg[/img] 广州佳途科技股份有限公司深知药物研发与质量控制的重要性,CATO标准品厂家,提供帕唑帕尼全套的杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展,以满足客户在药物研发和质量控制方面的需求。
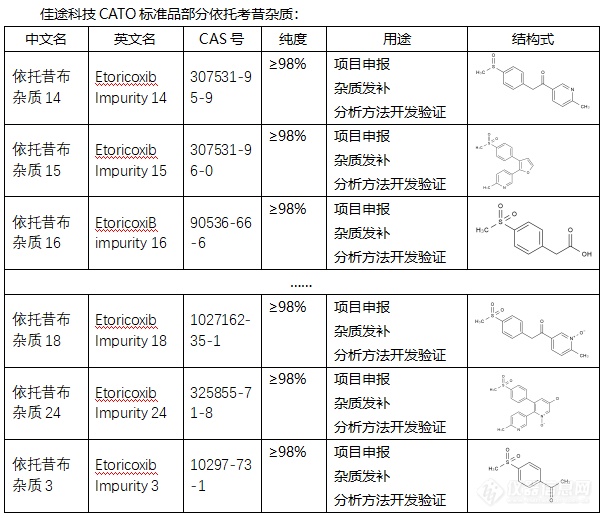
[font=宋体]◇依托考昔杂质[/font][font=宋体][font=宋体] 依托考昔杂质是在依托考昔的生产或保存过程中产生的非目标化合物。这些杂质可能会影响依托考昔的纯度和药效,因此在依托考昔的生产和质量控制过程中需要严格控制其含量。依托考昔杂质有多种类型,每一种都具有不同的化学特性,如[/font][font=Calibri]CAS[/font][font=宋体]号、分子式、分子量等。例如,依托考昔杂质[/font][font=Calibri]K[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]349536-41-0[/font][font=宋体],分子式为[/font][font=Calibri]C18H15ClN2O3S[/font][font=宋体]。依托考昔杂质[/font][font=Calibri]Etoricoxib[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]202409-33-4[/font][font=宋体],分子式为[/font][font=Calibri]C18H15N2O2SCl[/font][font=宋体],别名包括依托考昔、[/font][font=Calibri]5-[/font][font=宋体]氯[/font][font=Calibri]-2-(6-[/font][font=宋体]甲基吡啶[/font][font=Calibri]-3-[/font][font=宋体]基[/font][font=Calibri])-3-(4-[/font][font=宋体]甲基磺酰苯基[/font][font=Calibri])[/font][font=宋体]吡啶等。依托考昔杂质[/font][font=Calibri]Q[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]292067-97-1[/font][font=宋体],分子式为[/font][font=Calibri]C18H15ClN2S[/font][font=宋体]。此外,依托考昔还可能存在其他未具体命名的杂质。[/font][/font][font=宋体][font=Calibri] CATO[/font][font=宋体]标准品提供的依托考昔全套的杂质[/font][/font][font=宋体],[/font][font=宋体]这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分[/font][font=宋体]。[img=,604,518]https://ng1.17img.cn/bbsfiles/images/2024/02/202402182043373327_8351_6381607_3.png!w604x518.jpg[/img][/font][font=宋体][color=#05073b][back=#fdfdfe] 广州[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]佳途科技[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]股份有限公司[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]深知药物研发与质量控制的重要性[/back][/color][/font][font=宋体][font=宋体],[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供依托考昔全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展[/font][font=宋体],[/font][font=宋体]以满足客户在药物研发和质量控制方面的需求。[/font]
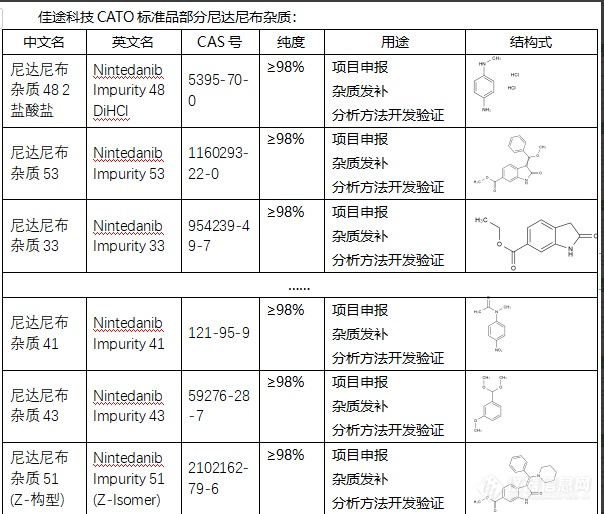
尼达尼布杂质可能会对药物的药理活性产生不利影响,导致药效降低。比如,有可能降低尼达尼布对肿瘤细胞的抑制作用,从而影响治疗效果。另一方面,尼达尼布杂质可能对人体产生毒性反应,对患者的身体健康产生不良影响。例如,有可能引起过敏反应,导致患者出现皮疹、呼吸困难等症状。但是,值得注意的是,这些展示都是可能性,并不一定在所有情况下都会发生。具体情况需要根据尼达尼布药品中杂质的类型和含量来判断。为了避免杂质对药物效果的影响,CATO标准品在药品生产中会进行严格的质量控制,对杂质进行有效的控制和清除。同时,掌握和理解杂质的产生机制,也有助于进一步完善和优化药品的生产工艺。[img=,604,514]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041439114552_8061_6381668_3.png!w604x514.jpg[/img]
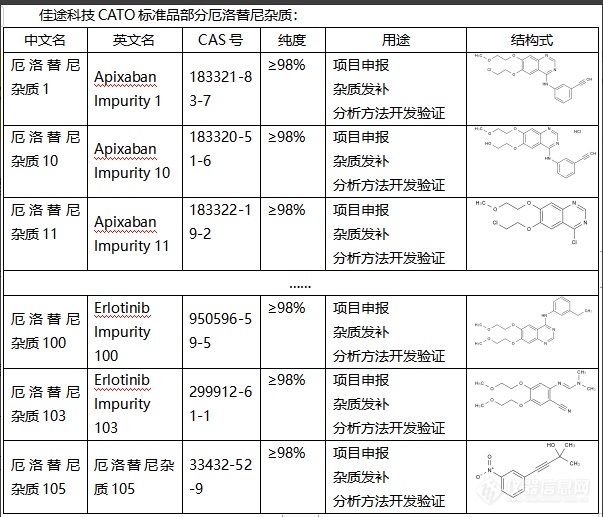
厄洛替尼杂质是药物制备过程中的副产品或污染物,可能会影响药物的安全性和有效性。常见的杂质有重金属、溶剂残留、有毒化合物等。对于厄洛替尼来说,杂质可能会影响其药效和安全性。一方面,杂质可能降低药物的纯度,从而降低其抑制肿瘤生长的效果。另一方面,某些杂质可能具有毒性或致敏性,可能导致患者出现不良反应。因此,对厄洛替尼的杂质进行检测和控制是药品质量控制的重要环节。这需要使用一种或多种分析方法,如高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法、[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法、质谱法等,以检测并定量药物中的杂质。CATO标准品针对可能的杂质源(如起始材料、反应条件、催化剂、溶剂等)进行控制,可以有效地减少杂质的生成。[img=,603,517]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021655361773_9862_6381668_3.png!w603x517.jpg[/img]
常用辅料及相关杂质在系统一时保留时间参考值极性色谱柱 辅料或杂质名称 相对保留时间 异丙醇 0.098 正丙醇 0.168 二甲基甲酰胺 0.500 乳酸乙酯 0.512 二甲基乙酰胺 0.591 二甲基亚砜 0.785 1,2-丙二醇 0.803 1,3-丁二醇 0.956 1,3-丙二醇 1.000 苯甲醇 1.083 二甘醇 1.178 肉豆寇酸异丙酯 1.206 三乙酸甘油酯 1.263 丙三醇 1.469 油酸乙酯 1.564 苯甲酸苄酯 1.683 中等极性色谱柱辅料或杂质名称 相对保留时间乙醇 0.281 异丙醇 0.333 丙三醇 1.000 三甘醇 1.227 以上是从别的地方看到的,有的样品出峰时间非常接近。在新药申报时遇到的问题是:当同一个样品在做检出限时,很可能出峰时间出现较小的变化,会不会影响到样品种类判断及资料申报呢?
[B][center]药品研发如何确定杂质限度[/center][/B][B]国家食品药品监督管理局药品审评中心 黄晓龙[/B] 在药品研发中,如何证实药品安全有效应该是研发人员始终关注的问题;而药品质量的稳定可控又是保证其安全有效的前提与基础。如果一个药品的质量不能达到稳定与可控,在使用时这一药品就不可能始终安全、有效,也就不能被批准上市。保证药品质量稳定可控,药品的纯度是一个重点。如何确定杂质的限度是药学研究人员与审评人员不能回避的关键问题,该限度的制订是否科学、合理,直接关系到药品的安全性与质量。药品在临床使用中产生的不良反应除与该药品本身的药理活性有关外,也有一部分与药品中所混入的其它杂质有关。例如,通过我国药学科技工作者数十年的努力,基本上确定青霉素等抗生素中的多聚物等高分子杂质是引起过敏的主要原因。所以在研发过程中一定要对药品中的杂质进行全面研究,并将杂质完全准确地控制在一个合理的范围之内。 尽管杂质限度的确定对于药品研发非常重要,但国内药品研发的现实情况并不令人乐观。从近几年的新药申报情况分析,在杂质的研究与限度确定方面存在着较多的问题,主要表现为:部分药品研究单位对杂质研究的重要性了解不深;标准中对杂质的控制不够全面与准确;制订杂质限度时考虑问题不够全面,很少考虑杂质对药品安全性的不良影响;即使在杂质的含量明显超出正常工艺所允许的范围时,也不注意对现有的处方与工艺进行必要的优化,以降低杂质的限度。◆杂质的分类 药品中的杂质一般分为三类:有机杂质、无机杂质及残留溶剂。 有机杂质是指在药品的生产与储存过程中产生的杂质,这些杂质可以是已知的、未知的、挥发性的或不挥发性的杂质,主要包括:降解产物、聚合物、原料药与辅料或内包材的反应产物、以及原料药制备过程中引入的起始原料、副产物、中间体、反应试剂、配位体与催化剂。由于这些杂质的化学结构与产品分子类似或具渊源关系,所以通常称之为有关物质。 无机杂质是指在药品的生产过程中产生的杂质,这些杂质通常是已知的,主要包括:反应试剂、配位体与催化剂、重金属或其它残留的金属、无机盐、过滤助剂、活性炭等其它物质。 残留溶剂是指在原料药及制剂的生产过程中使用的有机溶剂。 对于生产过程中引入的外来污染物,可通过“良好的生产规范”(GMP)来控制,故不属于本文所说的杂质范畴。原料药的不同晶型也不属于本文的讨论范畴。本文只谈有机杂质与无机杂质的限度确定。
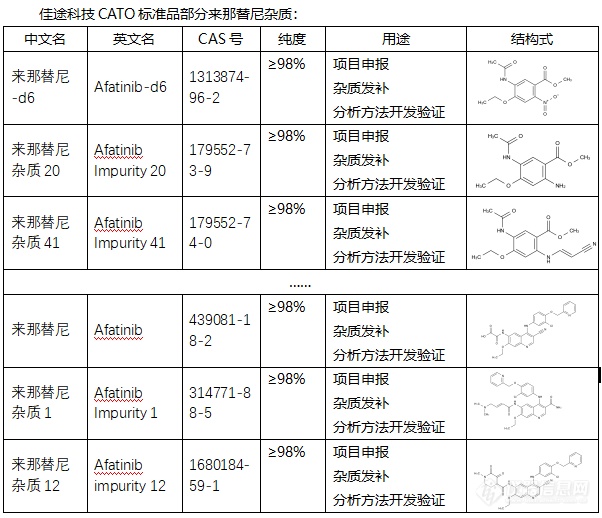
[font=宋体]◇来那替尼杂质[/font][font=宋体] 来那替尼杂质是在来那替尼药物制备或存储过程中可能产生的物质[/font][font=宋体],[/font][font=宋体][font=宋体]这些杂质可能会影响药物的纯度和效果,因此对其进行研究和控制对于确保药物的安全性和有效性至关重要。来那替尼杂质有多种,它们具有不同的[/font][font=Calibri]CAS[/font][font=宋体]号、化学式和分子量。例如,来那替尼杂质[/font][font=Calibri]NOQ[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]1348481-03-7[/font][font=宋体],纯度通常为[/font][font=Calibri]95% HPLC[/font][font=宋体]。此外,还有其他来那替尼杂质,如来那替尼杂质[/font][font=Calibri]1144516-15-3[/font][font=宋体]等。[/font][/font][font=宋体][font=Calibri] CATO[/font][font=宋体]标准品提供的来那替尼全套的杂质[/font][/font][font=宋体],[/font][font=宋体]这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分[/font][font=宋体]。[img=,602,518]https://ng1.17img.cn/bbsfiles/images/2024/02/202402192110007699_8786_6381607_3.png!w602x518.jpg[/img][/font][font=宋体][color=#05073b][back=#fdfdfe] 广州[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]佳途科技[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]股份有限公司[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]深知药物研发与质量控制的重要性[/back][/color][/font][font=宋体][font=宋体],[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供来那替尼全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展[/font][font=宋体],[/font][font=宋体]以满足客户在药物研发和质量控制方面的需求。[/font]
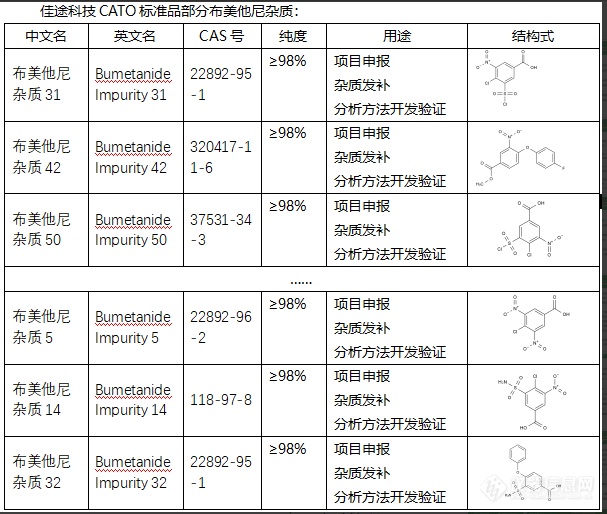
布美他尼杂质是药物布美他尼制剂中可能存在的杂质,这些杂质可能来源于原料药的生产过程、制剂的准备过程或者储存过程。1. 影响药效:药物中含有过多的杂质可能会影响药物的药效,产生药效不稳定、药效降低等问题。2. 影响安全性:部分杂质可能具有潜在的毒性,长期或高剂量使用可能对人体造成伤害。严重情况下可能出现药品不良反应甚至中毒。3. 影响药物的稳定性:不同类型的杂质结构,可能影响药物的稳定性,例如酸性或碱性杂质可能引起药物分解,导致药效降低或失效。4. 影响药物的外观质量:杂质可能会影响药物的外观性状,如颜色、透明度和溶解性等。因此,确定和控制药物中的杂质是药品质量控制的重要环节。CATO标准品对于药物杂质的研究,主要包括杂质的来源、形成机理、控制策略和杂质的鉴定、定量测定等内容。[img=,607,514]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041444015575_2473_6381668_3.png!w607x514.jpg[/img]
如题,最近接一个新项目,需要对其中一个基因毒性杂质建立可控的分析方法,该杂质是甲磺酸异丙酯,有开过类似方法的坛友吗?查了相关资料,欧洲药典有类似的GC方法,但我个人不想用GC,尤其是顶空进样,有没有好的方法?考虑过衍生,查到有人用2-巯基吡啶衍生,有做过这方面研究的同仁吗?请赐教,谢谢!
阿伐那非杂质是阿伐那非的同分异构体或相关化合物,其纯度、含量和杂质情况对阿伐那非的药效和安全性有重要影响。在药物研发和生产过程中,需要使用标准品来检测和鉴定阿伐那非及其杂质的性质和含量。COTO标准品是一种高纯度的标准物质,用于测定阿伐那非及其杂质的纯度、含量和化学性质。通过与COTO标准品进行对比和分析,可以确定阿伐那非及其杂质的结构、组成和含量,从而保证阿伐那非的质量和安全性。在药物研发和生产过程中,COTO标准品的使用非常重要。它可以提供可靠的参照物,用于质量控制、药物分析和化学计量学研究。通过使用COTO标准品,可以确保阿伐那非及其杂质的准确性和可靠性,为药物的安全性和有效性提供保障。总的来说,COTO标准品在阿伐那非杂质的研究和控制中具有重要作用。通过使用COTO标准品,可以更好地了解阿伐那非及其杂质的性质和含量,从而确保药物的安全和有效性。同时,也需要加强生产过程中的管理和监督,加强质量标准和监管措施的执行力度,确保药物质量和安全。
有机溶剂中杂质离子测试,蒸发时会干扰么?测的是碳酸二甲酯里面的杂质里金属离子,我用低温蒸发的办法,100度左右,想把溶剂除掉的同时保留杂质离子,然后水解进行测试。但担心这样溶剂挥发时会不会把大部分的金属离子带走,使得测得结果大大的偏低?谢谢!
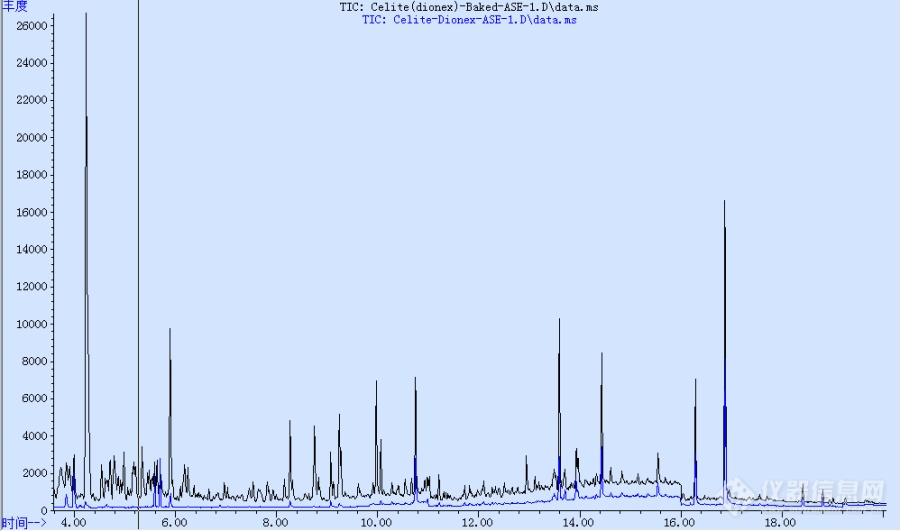
硅藻土和无水硫酸钠,分析纯,用马弗炉500度烘烤5个小时,冷却后使用。比较了烘烤前、后的空白实验,使用气-质的SIM模式,发现烘烤后的噪音大大增强了,百思不得其解。烘箱比较干净的,而且按道理说500度烘烤,有机干扰物应该都挥发掉了吧。测试的色谱条件:进样口300度,柱温最高295度.干扰是怎么带进去的?有没有可能烘烤过程中,把原来的一些比较惰性的杂质给分解了,杂质反而增多?[color=#FF0000][i][b]蓝色的为烘烤之前的硅藻土,黑色的为烘烤之后的。[/b][/i][/color][color=#FF0000][img=,690,407]http://ng1.17img.cn/bbsfiles/images/2017/10/201710310856_01_1870463_3.png!w690x407.jpg[/img][/color]
在做程序升温操作时,载气中的某些杂质,在低温时保留在色谱柱中,当拄温升高时不但引起基线漂移还可能在谱图上出现比较宽的"假峰"。7)仪器影响a.各类过滤器加速失效 b.调节阀(稳压阀,稳流阀,针形阀)被污染,气阻堵塞,调节精度降低或失灵;c.气路系统被污染,若要恢复仪器在高灵敏度情况下操做,有时要吹洗很长时间(可能一周以上)污染严重时有时再也无法恢复。d.检测器的寿命,实践表明,对ECD和TCD的寿命影响最明显,应引起用户特别注意。二.对气体纯度选择的一般原则1.从分析角度讲,微量分析比常量分析要求高,也就是说,气体中的杂质含量必须低于被分析组分的含量,如果用TCD分析10ppm的CO,则载气中的杂质总含量不得超过10ppm,因为99.999%纯度的气体则含0.001%的杂质,相当于10ppm所以对于10ppm的痕量分析,载气的纯度应高于99.999% 对于FID使用气体,碳氢化合物含量必须很低,载气中的大量氧杂质只要不对色谱柱造成影响,就不影响FID的性能,而操作ECD,载气中的氧气和水的含量必须很低等.2.毛细管柱分析比填充柱分析要求高 3.程序升温分析比恒定温度分析要求高 4.浓度型检测器比质量型检测器要求高 5.配有甲烷装置的FID比单FID操作的对载气中的微量CO,CO2要求要高的多.6.从仪器寿命和保持仪器的高灵敏度讲,中高档仪器比低当仪器要求高 三.操作不同检测器推荐使用的气体纯度我们推荐气体纯度的技术要求,通常用于常规分析,对于要求高灵敏度的痕量分析时,也可以使用更高纯度的气体。由于各个制气厂设置不同,其杂质含量将有所不同 为满足不同的使用要求,选用不同厂家不同纯度的气源后,可以通过气体净化处理满足分析要求。对于不同杂质的气体采用何种净化方法和装置有机会在加以讨论。综上所述,新购[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]接入气源时,一定要做到心中有数,决不能随意接入,否则会造成ECD,甲烷化装置等的损伤,信噪比减小的无法使用,下面给出了用于常规分析时,推荐使用的气体纯度(仅供参考):TCD 氦做载气:至少纯度为99.995%。杂质含量分别为:氖<10ppm 氮 <10ppm;氧<2.5 ppm 氩<0.1 ppm 二氧化碳<0.25 ppm。氢做载气: 至少纯度为99.995%。 杂质含量分别为: 氮<1 ppm 氧<5 ppm 二氧化碳<1 ppm 水<5 ppm 总烃<1 ppm 。FID 氮做载气: 至少纯度为99.998%。杂质含量分别为:氢<1 ppm 氧<1 ppm 氩<10ppm 二氧化碳<1 ppm 水<5 ppm 甲烷<1 ppm。氢气: 同TCD 空气: 呼吸级杂质:氩,氪,水,氦,氖均小于1%; 二氧化碳<500 ppm 一氧化碳<10ppm 总烃<0.02 ppm 甲烷<20 ppm。ECD 氮做载气: 至少纯度为99.998%。典型杂质同上。
因为换了工作,对现在公司的一些处理方式很不理解,问了一些同事,发现他们也不懂,只知道一直以来是按照那样用。可是我认为那样不对,遂发上来和大家讨论。以下说的都是我现在所在公司的做法:1.在进样之前,先进一针LOQ(定量限)溶液,如果LOQ中特定峰S/N(信躁比)=10或者连续进三针,峰面积RSD=10%(根据具体而定),则认为LOQ通过,否则不通过,不能进行后面的分析。2.在LOQ通过的情况下,进样品,然后用面积归一化法,对图谱进行分析,据此得出各杂质的含量,前面进的LOQ得到的数据不参与结果的计算。3.对于色谱图中,小峰的处理是这样的,峰面积百分比=LOQ(比如0.1%,将样品溶液稀释1000倍)则积上,否则不积。对于这样的做法,我主要有以下几个疑问:1.LOQ不是在做方法验证的时候才需要做吗?而且药典(包括USP)上明确写了,除了杂质定量外,都可以不做。那么每次都做个LOQ,有意义,有必要吗?2.将样品溶液稀释1000倍,得到的是浓度,怎么直接把它给转换成了峰面积比?这样科学吗?当然了,不考虑响应值的不同,即不引入校正因子。3.想问一下,大家对小峰是怎么处理的呢?一个小峰到底积不积上,根据什么来判断呢?我们以前基本是根据经验的。4.药典上不是说用面积归一化法对杂质进行定量分析是很不准确的吗?那仍然这样分析是否合理。5.药典上说的杂质定量都是与自身比对(与另一低浓度),那么我们这样的做法科学吗?希望大家不吝赐教!
[B][center]药物中杂质的来源及杂质限量检查[/center] [/B]药物只有合格品与不合格品;一般化学试剂分为4个等级(基准试剂、优级纯、分析纯、化学纯) [B]药物中一般杂质检查 [/B][B]氯化物为一指示性杂质。[/B] 通过对氯化物的控制,可同时控制与氯化物结合的一些阳离子以及某些同时生成的副产物。可从氯化物检查结果显示药物的纯度,间接考核生产、贮藏过程是否正常。 1. 原理 药物中微量的氯化物在硝酸酸性条件下与硝酸银反应,生成氯化银的胶体微粒而显白色浑浊,与一定量的标准氯化钠溶液在相同条件下产生的氯化银浑浊程度比较,判定供试品中氯化物是否符合限量规定。 Ag+ + Cl- → AgCl ↓ [B]硫酸盐检查法 [/B] 1. 原理 药物中微量的硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成硫酸钡的微粒而显白色浑浊,与一定量的标准硫酸钾溶液在相同条件下产生的硫酸钡浑浊程度比较,判定供试品中硫酸盐是否符合限量规定。 [B]铁盐检查法 [/B]硫氰酸盐法 巯基醋酸法 砷盐检查法 1. 古蔡氏法 1. 原理 金属锌与酸作用产生新生态的氢,与药物中微量砷盐反应生成具挥发性的砷化氢,遇溴化汞试纸产生黄色至棕色的砷斑,与同条件下一定量标准砷溶液所生成的砷比较斑,判断砷盐的含量。 [B]硒、氟及硫化物检查法 [/B]1. 氧瓶燃烧法 适用于以共价键结合的卤素、硫、硒的有机药物。 本法系将有机药物防入充满氧气的密闭燃烧瓶中进行燃烧,将燃烧所产生的欲测组分吸收于适当的吸收液中,然后根据欲测组分的性质,选用合适的分析方法进行鉴别、检查或含量测定。 [B]注意事项及讨论 [/B]1. 根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 2. 测定氟化物时应改用石英燃烧瓶。 1. 硒检查法 (1). 操作方法 样品与对照品液,调节Ph2.0±0.2,加盐酸羟胺,二氨基萘,比色。 [B]硫化物检查法 [/B] 方法同砷盐检查第一法,不装醋酸铅棉花,以醋酸铅试纸代替溴化汞试纸。 标准液取1ml 5/ml [B]澄清度检查法 [/B]将一定浓度的供试品溶液与浊度标准液分别置于配对的比浊用玻璃管,同置黑色背景上,在漫射光下观察。浊度标准液 硫酸肼与乌洛托品溶液混合分五个等级,未超过0.5等级即为澄清。BP98规定未超过1等级即为澄清。 [B]溶液颜色检查法 [/B]CHP2000 [B]1. 比色法[/B] 色调标准贮备液 黄色液 重铬酸钾液(BP98用氯化铁) 红色液 氯化钴液 蓝色液 硫酸铜液 配成各种色调色号标准比色液共50种。 [B]2. 分光光度法 [/B] [B]易碳化物检查法 [/B]检查药物中含有的遇硫酸易碳化或易氧化而呈色的有机杂质。 对照品液 样品液 加硫酸5后,加供试品。 [B]炽灼残渣检查法[/B] 取供试品1.0~2.0g或个药品项下规定的重量,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全碳化,放冷至室温;除另有规定外,加硫酸使湿润,低温加热至硫酸蒸气除尽后,在700~800炽灼使完全灰化,移至干燥器内,放冷至室温,精密称定,再在700~800炽灼至恒重,即得。残渣限量一般为0.1~0.2% 一般应使炽灼残渣量为1~2mg 若需将炽灼残渣留作重金属检查时,炽灼温度必须控制在500~600。 [B]干燥失重测定 [/B]1. 常压恒温干燥法 2. 干燥剂干燥法 3. 减压干燥法 [B]水分测定法 [/B][B]费休氏法 [/B] 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。 [B]甲苯法[/B] 在加热状态下,甲苯夹带着水分蒸出,收集蒸出的水分测定。 [B]药物中特殊杂质检查 [/B] [B]一、物理法 [/B] [B]二、化学反应法 [/B](一)容量分析法 (二)重量分析法 (三)比色法和比浊法 [B]三、色谱法 [/B]1.纸色谱法 薄层色谱法 TLC是药典中最常用的特殊杂质限量检查方法。 1.在一定供试品及检查条件下,不允许有杂质斑点存在 2.以待测杂质对照品检测 3.将供试品稀释到适当浓度作为杂质对照品溶液 4.选用质量符合规定的与供试品相同的药物作为杂质对照品 [B]高效液相色谱法 [/B] [B][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 [/B] 1.面积归一化法 2.主成分自身对照法 3.内标法测定 4.内标法加校正因子法 5.外标法 有机溶剂残留量测定法 [B]分光光度法 紫外分光光度法 比色法 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/B]
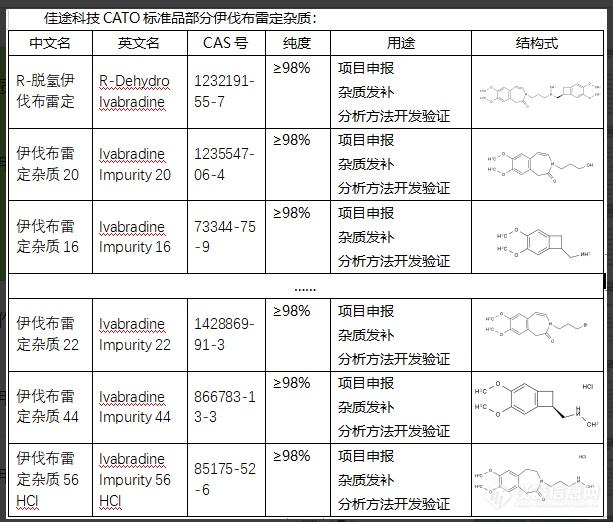
伊伐布雷定是一种用于治疗成人和儿童音乐综合症等疾病的药物。在伊伐布雷定的制造过程中,可能会产生一些杂质。这些伊伐布雷定杂质可能由化学反应生成,也可能来自原始材料。无论杂质来源如何,过多的杂质可能会影响药品的质量、疗效和安全性。例如,一些杂质可能会导致药物的疗效降低,或者引发不良反应。因此,制药公司必须在生产过程中严格检测和控制这些杂质,以确保药品的质量和安全性。CATO标准品对杂质进行研究和分析也可以有助于优化制药过程,例如,找出产生杂质的环节并进行调整,以减少杂质的生成。这有助于提高药品质量,保证疗效,保证患者的安全。[img=,613,522]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052104420028_3541_6381668_3.png!w613x522.jpg[/img]
我是一个新手,我做乙醇挥发性杂质时,我们的样品里面均未检出各杂质,是否合理?怎么感觉多少都应该有点呢?还是因为我是放置了几天以后才做样的缘故?请教各位!谢谢谢谢!
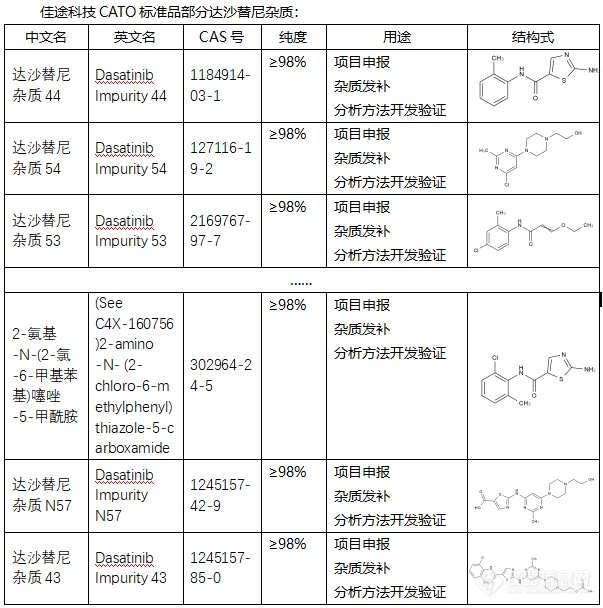
[font=宋体]◇达沙替尼[/font][font=宋体]杂质[/font][font=宋体] 达沙替尼[/font][font=宋体],[/font][font=宋体]其英文名为[/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]Dasatinib[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe],)是一种创新的第二代双重酪氨酸激酶抑制剂(TKI),也被称作DASA锡IB或商品名SPRYCEL(施达赛)。[/back][/color][/font][font=宋体]达沙替尼[/font][font=宋体][font=宋体]杂质过抑制[/font][font=Calibri]BCR-ABL[/font][font=宋体]蛋白的活性来发挥治疗作用。达沙替尼能够与[/font][font=Calibri]BCR-ABL[/font][font=宋体]蛋白结合并抑制其激酶活性,从而阻断白细胞的异常增殖,并促进正常白细胞的生成。[/font][/font][font=宋体][font=Calibri] CATO[/font][font=宋体]标准品提供的达沙替尼杂质用途主要是用于分析化学物质和质量控制的化学物质。[img=,603,608]https://ng1.17img.cn/bbsfiles/images/2024/02/202402062123365450_9441_6381607_3.png!w603x608.jpg[/img][/font][/font][font=宋体][font=宋体] 广州佳途科技股份有限公司,[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供达沙替尼全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,通过全面的检测、高效的沟通、专业的服务和完善的售后,确保所有产品均能现货供应[/font][font=宋体],[/font][font=宋体]致力于为客户提供高质量的产品和优质的服务,以满足客户在药物研发和质量控制方面的需求。[/font]
大家好,本人对DMF中杂质分析理解肤浅,希望大家一起探讨。杂质主要分为:1合成时产生的杂质 2 残留溶剂 3无机杂质wei616认为第2第3项容易做,ICH指南中已经有对溶剂的控制限度。无机杂质主要就是炽灼残渣和重金属,在相应标准中也有定量,所以都比较好做。关键是第1项不好做,我想问大家,该如何给合成时产生的杂质定量?这个标准该从哪里找?比如我在做一个试验A+B生成C,最终成品中A和B若有残留,限度该定多少呢?有相关的标准要求吗?谢谢大家了
广东化工杂志,现在改成半月刊,和广州化工杂志的页面几乎一模一样,很难辨别清楚,今年投了一篇论文,立马接收,然后立马缴费,等了很久之后打电话催促才寄发票,到付的。发票竟然是广告公司开具的发票,项目写成版面设计费,虽然广东化工单位开具了证明,但是本单位新规定不可以报销,必须是这个杂志或者广东省广远石化集团有限公司或者广东省石油化工研究院开具的发票,打电话咨询杂志社,杂志社说:是挂靠单位,与上述两个公司没有任何财务往来,独立经营,自负盈亏,是挂靠在这两个单位名下的,杂志社本身没有开具发票的资格。唉。不能报销,我只能自己买单了。在此,提醒各位,发文章还是要好好咨询一下才行的。
2005年3月18日国家食品药品监督管理局注册司正式发布了2003年开始起草的化学药16个技术指导原则。为配合这些指导原则的实施,有必要采用各种方式对这些指导原则进行宣传,以加深各有关方面对指导原则内涵的理解。根据在《杂质研究的技术指导原则》的起草过程中及发布后所收集的意见进行分析,发现不少同仁对其中的一些内容尚不太理解,作为主要起草人,我想谈一些个人的看法,以供大家在学习使用该指导原则时参考。 1. 质控限度如何使用的问题 在该指导原则中,根据原料药与制剂的临床用药量的大小分别提出了单个杂质的质控限度。但在进行质量研究与标准制订时如何使用这些限度,尚存在一些疑义。现对该限度的使用解释如下:在本指导原则中反复强调,在制订质量标准中的杂质限度时需首先考虑杂质的安全性。而杂质的安全性评价主要是综合药物研发过程中所进行的药理毒理及临床研究中的毒副作用与不良反应来判断的。由于上述研究所用的样品本身会包含一定种类与数量的杂质,所以如果在这些研究中并未明显反映出与杂质有关的毒副作用,则可认为该杂质已经通过了安全性的验证。在此前提之下,如果该杂质的含量同时也在正常的制备工艺所允许的限度范围内,那么根据试验样品中杂质的含量所确定的限度可认为是合理的。之所以引入质控限度的规定,主要是为了减少对杂质安全性的验证。如果某一杂质的含量低于规定的质控限度,则不必对该杂质的安全性进行确认,除非已知该杂质具有明显的毒性。 2. 附件中为何不设定总杂质限度的问题 在指导原则中提出的各种限度都是针对单个杂质的限度。之所以未规定总杂质的限度,主要是考虑:就每一个具体的药品而言,所含杂质种类的多寡不一,每个杂质的含量也不尽相同,因此,很难在指导原则中给出一个统一的总杂质限度。尽管指导原则中未规定总杂质的具体限度,但是作为杂质控制的一个重要参数,指导原则中已明确要求分别制订每一个已知杂质、未知杂质及总杂质的限度。 3. 为何设定鉴定限度的问题 在国外相关的指导原则中,规定对超过鉴定限度的杂质都要研究鉴定该杂质的化学结构。我们在起草本指导原则时,根据目前国内药品研发的实际水平,也曾考虑过是否有必要在此引入鉴定限度的要求。经过与各方面专家、研发人员反复讨论,最终决定引入鉴定限度的概念,在指导原则中要求:“对于超过鉴定限度的杂质应作进一步的研究,确定其来源,推测其可能的结构,进而判断该杂质对药物安全性的影响;对于在稳定性研究中产生的超过鉴定限度的降解产物也应做相应的研究。对于未超过鉴定限度的杂质一般不需进行结构研究。对于可能具有特殊的生理活性或毒性的杂质,则应进行结构确证和安全性验证。” 当时决定引入这一限度的一个基本考虑是因为杂质的毒性与其化学结构密切相关。对于含量较大的未知杂质,如果能通过一系列的药学研究确定其化学结构,则可利用已有的结构-毒性数据库,初步预测该杂质的毒性,从而为该杂质限度的制订提供依据,以充分保证患者用药的安全性。例如,法国药政部门就规定:对于可能具有致畸毒性的杂质,其每天摄入的剂量不得过1.5ug;对于有致畸毒性的杂质,其每天摄入的剂量不得过0.15ug,限度的要求更为严格。而这些规定都是基于杂质的结构已知基础之上的。总之,随着我国药品研发水平及人民生活水平的逐渐提高,对药品质量的要求也会不断提高。作为一个有远见、负责任的药品研发与生产单位,一定要顺潮流而动,不断为国人提供更多更好的药品,其自身也才能不断地发展壮大。
化药质量控制CTD申报资料中杂质研究的几个问题张哲峰成海平宁黎丽田洁化药药学二部 摘要:杂质研究与控制是把控药品质量风险的重要内容之一,基于杂质谱分析的杂质控制是“质量源于设计”基本理念在杂质研究与控制中的具体实践,需要与CMC各项研究乃至药理毒理及临床安全性研究等环节关联思考、综合考虑,而不仅仅拘泥于提供准确的分析数据。本文针对当前CTD申报资料中杂质研究方面存在的问题与不足,结合CTD过程控制和终点控制相结合、研究和验证相结合、全面系统的药品质量控制理念,探讨仿制药杂质研究与控制的基本逻辑思路,提出CTD申报资料中杂质研究与控制方面几个需要关注的问题。 关键词:杂质研究与控制 杂质谱 CTD格式 杂质研究与控制是一项系统工程,需要以杂质谱分析为主线,安全性为核心,按照风险控制的策略,将杂质研究与CMC各项研究,乃至药理毒理及临床安全性研究等环节关联思考、综合考虑,而不仅仅拘泥于提供准确分析数据的传统思维,不是一项孤立的分析工作。CTD(Common Technical Document)申报格式体现了过程控制和终点控制相结合、研究和验证相结合、全面系统的药品质量控制理念,更加符合杂质研究与控制的基本规律和逻辑思路。自2011年4月起,药审中心陆续发布了多项有关CTD格式及技术审评的相关要求及电子刊物,对于国内研发单位正确理解CTD格式内含的基本精神起到了一定的促进作用,但就目前阶段的申报情况看,有些申报资料在杂质研究方面仍存在一些不足,仅仅是形式上的CTD格式,尚未实质性贯彻CTD的基本逻辑思路。以下是针对目前CTD申报资料中杂质研究相关问题的一些考虑。 1、CTD格式中杂质控制的考虑要体现在CMC的各个环节,而不是仅仅局限在“质量控制”模块。如制剂的原辅料控制中,原辅料的选择与控制要考虑以符合制剂质量要求(杂质等)为核心,必要时进行精制处理并制定内控标准;关键工艺步骤及参数的确立、工艺开发过程等要考虑以杂质是否得到有效控制为重点关注之一;制剂相关特性中要体现与原研产品杂质谱等的对比情况;包材、贮藏条件以及有效期的确立等也要以杂质是否处于安全合理的可控范围内为核心等等。实际上这正是源头控制、过程控制与终点控制相结合的杂质控制理念的体现,在研发工作及申报资料的整理中都需要针对性的贯彻实施。 问题与案例:有些申报资料在某种程度上未能充分体现杂质研究的整体性,对杂质控制措施仅强调了终点控制措施,尚未充分体现源头控制与过程控制的基本思路,具体表现在如下方面: (1)制剂杂质控制受制于原料药质控水平的约束,以目前国内批准的原料药杂质水平现状为由,未能根据该品当前杂质控制的水平与趋势,对原料药提出较为严格的针对性的杂质控制要求,并进行质量内控,因而难以确保制剂杂质控制水平与目前国际水平相适应; (2)在论述说明制剂相关特性时,未提供与原研产品杂质谱的对比分析情况; (3)关键工艺步骤及参数的确立、工艺开发过程相关内容中未详细说明杂质谱的变化情况,缺失关键质量数据的支持。 2、CTD格式的特点之一是研究内容模块化呈现,但需关注杂质分析与控制的系统性与整体性,不能割裂各项内容的必然联系和有机统一。比如对原料药而言,杂质分析与控制的相关内容会分布在分析方法(3.2.S.4.2)、方法学验证(3.2.S.4.3)、杂质对比研究与杂质谱分析(3.2.S.4.5)、杂质情况分析总结(3.2.S.3.2)、样品检测与数据积累(3.2.S.4.4)、控制限度(2.3.S.4.1)等各模块中,但杂质研究又是一项系统工程,具有统一的整体性,因此,不要因为申报资料格式的模块化而人为割裂各项研究内容的相互联系,甚至遗漏相关研究内容,要高度关注杂质分析与控制的系统性与整体性,将杂质研究与控制的全部内容和信息体现在相应模块中。如详细的杂质研究报告可以体现在3.2.S.4.5中;3.2.S.3.2要报告杂质研究的结果;杂质分析方法的筛选、研究与验证内容要在3.2.S.4.3中体现;对仿制药而言,杂质限度确定的论证与依据需要在与原研产品进行全面的杂质谱对比研究基础上进行论证说明,因此,与原研产品的对比研究及结论要在3.2.S.4.5中体现。 问题与案例:有些申报资料忽视了各项研究间的关联性,未能充分突出仿制药研发的特点,具体体现如下: (1)对分析方法的来源没有足够明确的说明,在分析方法的筛选、优化等方面做了哪些研究等信息缺失,只有方法学验证内容,缺失方法学筛选研究的相关信息; (2)没有比较说明采用的杂质分析方法与USP、BP/EP、JP等药典同品种分析方法相比有哪些区别和优势,未能从分析方法、杂质控制种类及限度要求等方面的比较情况评估拟定质量标准的杂质控制水平; (3)缺失放行标准质控限度确定的依据进行论述,3.2.S.4.5中只对货架期标准限度的确定进行了相关说明,未从杂质来源与特点、数据积累、稳定性考察等角度论述放行标准中相关限度确定的依据。 3、从杂质谱分析入手确立科学的杂质研究基本思路。杂质谱包括药物中所有杂质的种类、来源及特性等信息,通过杂质谱分析较为全面地掌握产品中杂质概貌(种类、含量、来源和结构等);有针对性地选择合适的杂质分析方法,以确保杂质的有效检出和确认;通过与原研产品杂质谱的对比研究,根据各相应杂质的一致性求证,或跟踪杂质谱对安全性试验或临床试验结果产生的影响,评估各杂质的安全性风险和可接受水平;结合规模化生产时杂质谱的变化情况,确立安全合理的杂质控制水平。 基于杂质谱分析的杂质研究是一种“以源为始”的主动思维模式,以“质量源于设计”的观点,从杂质来源入手,从制备工艺、化学结构、处方组成的分析出发,评估、预测产品中可能存在的及潜在的副产物、中间体、降解物以及试剂、催化剂残留等大体的杂质概况,辅以适当的强制降解、对照物质的加入等验证的手段,考证建立的分析方法是否能够将它们逐一检出,并进行相应的方法学验证工作;相比之下,传统的杂质研究是一种“以终为始”的被动行为和逆向思维模式,从杂质分析的结果出发,仅从建立的某种检测方法所检出的有关物质中归属其来源情况,而未充分分析与验证可能存在的潜在杂质情况,建立的分析方法能否全面检出这些杂质,故容易出现杂质漏检的情况,难以全面掌握产品的杂质谱。 问题与案例:杂质谱分析表明某原料药所采用的合成路线会产生具有遗传毒性的双叠氮杂质ROX,但最初建立的分析方法未检出该杂质,但不能确定是产品中的确不存在该杂质,还是建立的分析方法不能有效检出该杂质,经定向制备该杂质,采用标准加入法有针对性的分析考查,采用改进后分析方法在正常产品中检测到该物质,尽管其含量极低,考虑到其较强的毒性情况,质量标准中仍作为特定杂质予以严格控制,保证了其临床应用的安全性。因此,基于杂质谱分析的杂质研究是一种相对科学的思维模式,对于有效掌控杂质安全性风险具有重要意义。 4、分析方法的验证应具备针对性和全面性。杂质的微量性和复杂性,使得检测方法的专属性、灵敏度和准确度十分关键。杂质分析方法的对象是各个潜在的杂质,因此,其分析方法的验证需要根据不同杂质的特点综合设计验证方案,进行有针对性的规范验证。 问题与案例:一些申报资料中对杂质分析方法的灵敏度、准确度等仅仅针对主药化合物进行验证,仍无法说明是否适用于相关杂质的检出和定量。如下是某药物有关物质测定方法学验证总结,基本体现了方法学验证的针对性与全面性,建议参考。 项目验证结果专属性①统适用性良好,峰形、峰纯度、柱效参数等符合要求。②在酸、碱、高温、氧化各破坏条件下的主峰峰纯度角均小于纯度阈值,主峰峰纯度良好,破坏后的主峰与各杂质峰均能有效分离,分离度均大于1.5。③供试液加入起始原料、反应试剂、副产物及中间体等,均可有效检出,并良好分离。线性和范围①质A浓度在0.036μg/ml~1.204μg/ml范围内,线性关系良好(n=8);y=13703x-174.83;R2=0.9991。②杂质B浓度在0.089μg/ml~1.192μg/ml范围内,线性关系良好(n=7);y=10941x-517;R2=0.9995。③杂质C浓度在0.299μg/ml~4.784μg/ml范围内,线性关系良好(n=7);y=13257x-492.44;R2=0.9999。④主药浓度在0.3μg/ml~4.8μg/ml范围内,线性关系良好(n=6);y=15008x+565.48;R2=0.9999。灵敏度杂质A、B、C及主药最低检出限分别为0.012ng、0.0

大家在冲调奶粉的时候,很少有观察里面杂质的吧。如果不细致的观察,细小的颗粒里面 杂质很难看到。今天在验收一批全脂粉原料的时候,就发现了大量的杂质。晒出来给大家看一下。大家讨论一下 奶粉中的杂质是从哪里来的呢? http://ng1.17img.cn/bbsfiles/images/2014/02/201402191858_490605_2227357_3.jpg
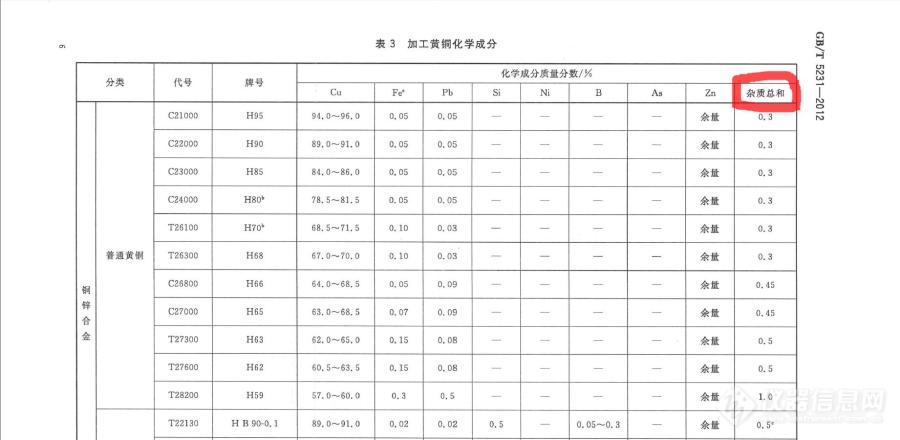
[img=,690,337]https://ng1.17img.cn/bbsfiles/images/2019/06/201906141645148522_8346_1925586_3.jpg!w690x337.jpg[/img]最近在接到个黄铜的样品,需要判定。看标准发现有个杂质总和的项目。[img=,690,76]https://ng1.17img.cn/bbsfiles/images/2019/06/201906141648248781_1429_1925586_3.jpg!w690x76.jpg[/img]这个杂质总和究竟是指只有具体限量值的,还是包括打横杠的元素呢?
大概除了一些刚入门的学生外,很多人都同意,根据一个人在顶级杂志比如Nature 或Science 杂志上发表文章的数量不是决定一个科学家优劣的唯一标准。但是,在这些杂志上发表文章,相信仍然是许多人梦寐以求的。那么在“顶级杂志”上发表论文为什么就这么让人趋之若鹫呢? 第一,你的实验结果或学术观点会得到极其迅速的传播。Nature和Science是科学界“老少咸宜”的读物。你的观点通常会被其他或相近领域的人迅速的接受(相对与其他low profile杂志),你也会从他们那里得到很多措辞含糊的赞扬。而许多你自己领域的研究人员会“恶毒“的批评你。 第二,在 Nature和Science上发表文章,你的结果会被认为具有“普遍意义”。 第三,你和你的graduate students或postdoctoral fellows 会得到比长效玛啡注射更强的激励效果。 第四,那些原来已经拒绝你的graduate student or postdoctoral fellow的工作申请的学校,会打电话给你,说要重新考虑他或她的申请。而你会翘着脚,对著电话说“Well, 嗯...,让我再想想....”。 第五,你的department chairman在公开场合,仍然会拍着你的肩膀,说一些可有可无的话,但最高级形容词的用量会有明显增加。
和大家谈一些怎么发中药的文章,做中药的在中国那是多了去了,号称是五千年精华嘛。谁研究谁爱国一样的来了很多人。熬点药水,灌灌老鼠,称一下肿瘤,发现轻了很多。其实是因为灌药灌的老鼠难受了,不吃不喝饿的。这样的研究数据可信度比较低,所以适合发最低的一些SCI 杂志,勉强凑合发了就算了。AJPP 还是可以发的,没人查你发的是真是假。连方肘子也忙不过来——周发几十篇。进步一点的工作,是把中药提出来,化学方法,但不怎么纯化。可以扔到肿瘤细胞培养皿里,过几天数数,发现细胞死不少。做几个蛋白实验,发现caspase-3出来了。不错,细胞凋亡了。也有人说是加啥冷的液体细胞都会死,所以只要加药就能出文章。这类工作发的杂志好一些,毕竟有分子分析,可以上个MMP,MMR,IJMR啥的。再进步一点的工作,中药被取成为 “一类混合物”,比如 枸杞多糖,人参皂甙,不管有多少种,大概的生理活性是可类比的,这样的话就可以冲 1-2分的文章了,无论是做细胞,还是做在体动物模型,效果好可以发更好的文章。这类工作就可以发各个领域专门的杂志了,比如 Cancer research, Int Orp, ..等等。又进一步,就是2-3种以下的混合物 以及单体。 BJP 就是这个要求。或者 BCJP成分有了,分子式你最好也提供一下。拿单体来做实验基本说啥就是啥。这样的工作我看以前可以发 PNAS, 现在大多数在 Plos One 附近。当然这些是中药提取物。 其他的一些植物性提取物(比如 咖喱里的)的一些成分,做出来的文章发了Nature 子刊,以及十几分的刊物上,还是有很多的。这个,是怎么做的问题。那么,该评职称了,该毕业了,该申请经费了,该拿课题了,得发文章。如何发呢?我个人的一些经验(读过的其他人的论文,修改的,经手的)。首先写清楚是啥药。这药以前能干啥,现在你干了啥。是混合物还是单体。分别的生物/医学活性是什么。中药文章很多人一写就写补肾阳调整经络。这个老外不懂,你得写,在酒精中毒模型中,对肝细胞有保护作用,通过 PI3K/Akt 通路。这个一写出来,你又做了一个保护大脑的实验,老外觉得,不错不错,make sense印象就不错。逻辑通出来了。只要有一个逻辑对了,这个文章基本就靠谱了。有分子结构的稍微提供下。浓度弄清楚。IC50列出来。其次是代谢了这药还是这药不。混合物过一下肠子吸收了再循环两圈 可能大变样。讨论里写清楚,不然对有效成分弄不好,别人觉得你不懂这药。再次是把中药当成西药写。特别是使用方法,剂量,如何控制扩散什么的。不然老外也搞不明白为啥这样给药,和女巫一样。还有讨论里不要大谈混合物。多谈谈意义,这药这样了,它的活性成分这样了,有奇效。原因是啥,结合在哪个受体什么的。重心从中药转移到细胞/动物的变化上,老外就忘掉这个是混合物了。参考文献上尽量少引用中文。我不是说不引,10个里面有1个还是能接受的。中药有太多东西,让你写文章时 很想引中文文献。但是要控制。这个现在中国人帮中国人审稿最爱挑剔这个。不评价。还有就是,去哪发?BJP,这种非常严格的杂志,发的非常少。混合物他们觉得不靠谱,大家谨慎就是。我统计了一下我们团队以前处理的稿件,大部分发在专科的医学,和综合性药理学杂志上。我个人认为综合性的医学杂志不太爱收中药稿件,因为他们可以选择发的东西很多。中药就显不是那么牛比。而综合药理学杂志则觉得中药治百病,各类都可以上。还有一些非常专业的杂志如果你有很好的治疗效果,也可以发过去。同时,Plos One 现在是3分以上发中药文章的大户,我非常建议大家尝试。
在有关物质方法开发过程中,会考察一些杂质,与主成分混成一个系统适用性溶液,在同一波长下检测,但有些杂质可能与主成分响应不一致,就需要引入校正因子,但是本人没有经过这方面的培训和学习,对这方面的规范操作不是很明白,希望有这方面经验的老师和同仁们,不吝赐教。问题:1、对于考察的杂质而言,方法验证时,是所有杂质都需要做校正因子,还是有选择的?比如只选择与主成分响应不一致的杂质进样校正因子测定?2、如何判断杂质与主成分响应值不一致?是在DAD检测器下的图谱中,看峰的紫外扫描图吗?3、杂质校正因子与相对校正因子的计算公式?我们目前是将已知杂质的两个浓度(一个是约0.1mg/ml浓度的,另一个是0.01mg/ml)各进3针,取平均值,校正因子=峰面积/浓度;相对校正因子=杂质校正因子/主成分的校正因子。







 我要推广仪器
我要推广仪器
 下载APP
下载APP