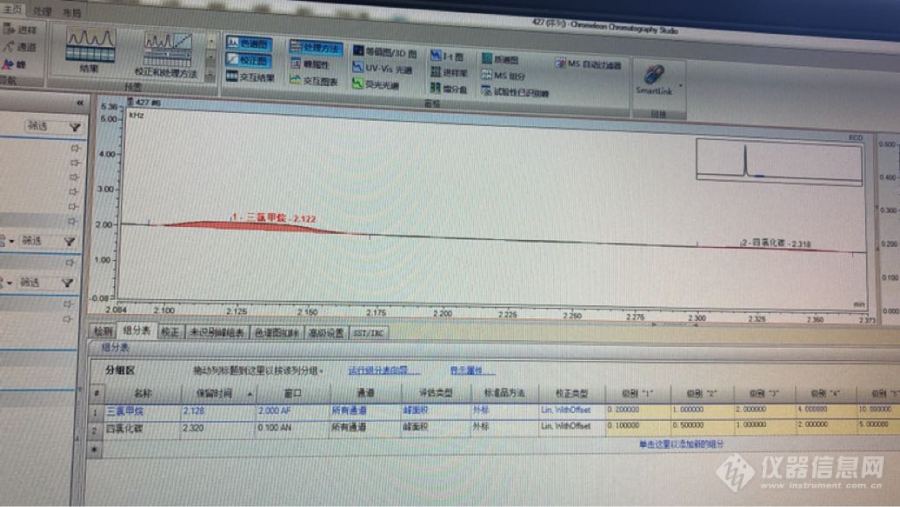
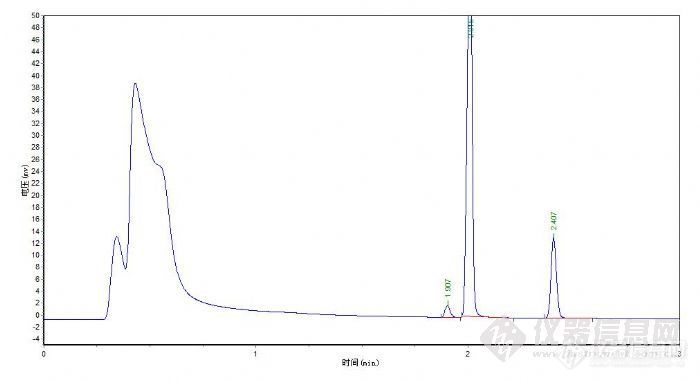
检测自来水中三氯甲烷和四氯化碳,三氯甲烷有峰 但是结果显示na,四氯化碳峰比三氯甲烷的小 但是有数值结果[img=,690,388]http://ng1.17img.cn/bbsfiles/images/2018/05/201805041032060962_2993_3400610_3.png[/img][img=,690,516]http://ng1.17img.cn/bbsfiles/images/2018/05/201805041032074701_6475_3400610_3.png[/img]
三氯甲烷与二氯甲烷的区别
最近要用气质考核这个三氯甲烷和四氯化碳,给的浓度范围是三氯甲烷是100-200mg/L,四氯化碳是30-80mg/L,我应该是把线陪在未知样周围,对吧但是对于混标以前没做过,所以有些疑问,请各位高手帮忙解释一下,先谢谢了一、怎么能把标液的浓度分别均为1000mg/L的三氯甲烷和四氯化碳配成混标? 现在有个棘手的问题,没有那么小的容量瓶来定容。我设定的是分别取0.75mL的三氯甲烷和0.24mL的四氯化碳定容在3mL,可是没有这么小的啊二、安捷伦的自动进样瓶可以作为定容的容器吗?先想到这里,希望没睡的兄弟姐妹们帮帮忙吧
二氯甲烷和三氯甲烷的区别
方法:取本品约1.0g,精密称定,置顶空瓶中,精密加水5ml使溶解,密封,作为供试品溶液。精密称取乙腈、丙酮、二氯甲烷适量,用水定量稀释制成每1ml约含乙腈0.41mg、含丙酮5mg、含二氯甲烷0.6mg的混合溶液,精密量取5ml,置顶空瓶中,密封,作为对照品溶液,照残留溶剂测定法(中国药典2005年版二部附录Ⅷ P)测定。以5%苯基-95%二甲基聚硅氧烷(SE-54)(或极性相近)为固定液的毛细管柱为色谱柱,柱温为400C,维持12分钟,进样口温度为2000C,检测器温度为2500C;顶空瓶平衡温度为800C,平衡时间为30分钟。取对照溶液顶空进样,计算数次结果,其相对标准偏差不得过5%。取供试品溶液与对对照品溶液分别顶空进样,记录色谱图,按外标法以峰面积计算,含二氯甲烷不得过0.06%,含乙腈量不得过0.041%。含丙酮不得过0.5%。http://ng1.17img.cn/bbsfiles/images/2011/11/201111011450_327697_2209208_3.jpg检测器用的是FID检测器,出峰顺序为:乙腈,丙酮、二氧甲烷。请各位版友分析一下这是什么原因?1. 在1分钟前出的怪峰,是不是因为顶空瓶压力跟载气上的压力调节得不一致的原因。混合对照时出怪峰,单独进水时不出怪峰?2. 为什么浓度相同的二氯甲烷和乙腈,反倒用顶空进样时,乙脯的峰比二氯甲烷小10倍左右?
前段时间,俺采用液体自动进样塔测定,以直接进样的方式测定溶剂残留,残留为三氯甲烷,采用ECD检测器,遇到了进样间隔残留量非常大的问题,然后请教了大家。大家给俺出了很多主意,最统一的就是俺的进样量太大了,俺的直接进样量是0.05mg/ml;这一次俺将进样量降至了0.5μg/ml, 进样残留间隔果然减小了;然后俺发现,如果将色谱柱温度(俺用DB-624)升至85摄氏度,高于三氯甲烷的沸点,进样残留间隔也可以大大减小。所以俺总结,如果需要减小直接进样的残留量,可以考虑1 直接减小进样量,2 在分析条件允许的前提下,适当升高色谱柱温度。再次感谢大家的帮助。
求助!!小弟最近有一个品种,测定二氯甲烷和三氯甲烷,采用ECD做,样品是水溶性,所以我采取的方法如下对照品溶液,先用DMF溶解,再用水稀释至相应浓度仪器方法:顶空 温度对应是 70、80、90℃,平衡15分钟 程序升温,,40保持3分钟,15的速率到180保持3分钟,进样口200,检测器300℃二氯甲烷,三氯甲烷依次出峰,问题来了,二氯甲烷线性不错,但是三氯甲烷根本不成线性(浓度范围从 0.003ug/ml---0.06ug/ml)第一个点峰面积 3000,第二个点不到4000,第三个点3100多点,第四个点6000多,第5个点不到9000。。。。
假设同等浓度的一氯甲烷和二氯甲烷,用FID检测器检测,那么一氯甲烷的峰面积大还是二氯甲烷的峰面积大?那个大虾知道?
近期在测试二氯甲烷水分时 发现水分测试结果有越测越高的现象,排除仪器参数的问题,电量法和容量法测试都是这样的。我是用塑料针管进的样品,不知道是不是与这个有关系。另外,样品的水分非常之小0.01%以下。还是和环境的温湿度有关系?
卤代烃对一般有机分子的溶解性能都很好,为什么在进行核磁测试的时候,一般选用三氯甲烷而不是二氯甲烷(氘代)。另外,乙酸乙酯一般也不用于核磁的溶剂,为什么?谢谢。
三氯甲烷和二氯甲烷哪个毒
三氯甲烷(Trichloromethane),化学式为CHCl?,是一种有机化合物,也被称为氯仿。它可以通过甲烷和氯气在光照条件下的反应制得。具体反应过程如下:? ?甲烷和氯气在光照条件下的反应?: 甲烷和氯气在光照条件下发生取代反应,逐步取代甲烷分子中的氢原子,最终生成三氯甲烷。反应过程如下:(CH_4 + Cl_2 \rightarrow CH_3Cl + HCl)(CH_3Cl + Cl_2 \rightarrow CH_2Cl_2 + HCl)(CH_2Cl_2 + Cl_2 \rightarrow CHCl_3 + HCl)(CHCl_3 + Cl_2 \rightarrow CCl_4 + HCl) ?三氯甲烷的其他生成途径?: 在有机合成中,三氯甲烷是重要的原料,生产或使用三氯甲烷时的意外事故可能导致环境污染。大气中的三氯甲烷部分是由于三氯乙烯在光化作用下降解而成。在饮用水处理中,使用氯消毒时,某些有机氯化合物(主要为三氯甲烷)可能形成,其含量高时可能对健康产生危害。
二氯甲烷和三氯甲烷哪个毒性大
生活饮用水检测三氯甲烷的结果和余氯含量是否有正相关的关系?或者说不含余氯,就一定不存在三氯甲烷,还是有三氯甲烷就一定有余氯?
《GBT 20388-2006 纺织品 邻苯二甲酸酯的测定》中规定萃取试剂和配标准品的试剂都是用三氯甲烷,但考虑到三氯甲烷毒性太大,能都用二氯甲烷代替,有做过的版友么?还有如果萃取一定要用三氯甲烷的话,配制标准品是否能用二氯甲烷?
客户做药里面的东西,用二氯甲烷做空白,242nm处,但校零后波动较大,是什么原因呢?如果用242nm测水就比较稳定,二氯甲烷这个样品有什么特殊的呢?请大家指教
浓度1000ppm左右的二氯甲烷出峰面积只有5000左右,基线上一个很小的一个突起。进气量250微升,除了提升进样量之外还有没有别的办法让峰面积更大一些?
大家都知到三氯甲烷是要备案才能买的。硝酸银也是要备案才能买的但是如果买成品硝酸银溶液就可以避开备案的要求了。。所以我反向思考: 买配制好的 (三氯甲烷冰乙酸混合液),是否可以成功避开备案的要求呢?? 如果需要的话,,
请教:在实验室的基本条件下,能否对氯甲烷进行处理,使Cl和-CH3脱离(话比较白,见笑),最好能够使之在两种不同的物质中存在,容易操作,并且容易进行分离。看了一些资料,说卤代烷和硝酸银可以反应生成沉淀,不知道跟氯甲烷的反应如何?容易程度如何?谢谢
请教关于原料氯甲烷的问题:1.盛氯甲烷的钢瓶用什么减压阀减压(有氯甲烷专用减压阀吗)? 2.检测氯甲烷纯度用FID检测器(面积归一定量)行吗?3.如果要送到外单位检测,如何取样啊?请推荐几个可分析氯甲烷纯度的检测单位吧。谢谢
正常条件下,氯甲烷和溴甲烷都是气态,请问如何称量配制氯甲烷和溴甲烷标准溶液?不想买现成的标液~求指导~
看到GB用二氯甲烷提取果汁中农残,为啥呢?怎么不用乙腈呢?实验室没有二氯甲烷可以用别的代替吗?算不算方法偏离?
ECD用624的柱子空气中的氯甲烷响应值很低,怎么办?
赛默飞[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]顶空法测三氯甲烷四氯化碳,响应值很小,怎么调大呢?最左边应该是空气峰,中间还有一个很大的峰,是甲醇峰吗?搞不懂
二氯甲烷、三氯甲烷、三氯乙烯和1,4-二氧六环浓度(ug/ml)分别为12,1.2,1.6,4.6。顶空进样。 G1888的顶空进样器,80度平衡60min,再进样1min。 6850GC,柱子是AT-624,30m*0.53mm*0.32um,起始柱温40,保持20min;再80/min到240,保持20min。进样口温度是250,检测器是250.载气是氮气。横流模式,流速1ml/min。 但是对照响应非常小,估计信噪比只有2~3. 在方法运行前,发现柱头压很低,只有0.7psi,一般都是6左右。 对照是用水溶解的,开始发现有不溶物,呈油珠,后来溶解。 请大家指点下,有可能是什么原因呢?
客户购买公司次氯酸钠,结果出厂水三氯甲烷含量高,就怀疑我们的次氯酸钠含有三氯甲烷,按理想来说是不应该有的,结果他测出来了又,而且较高,但我们送到两家第三方机构去检测,结果是有,但是没比客户测出的低差不多百倍,所以想买三氯甲烷标样给几个检测单位,不知道买什么样的样品。(哪位大侠也可和我讨论下次氯酸钠中三氯甲烷的来源)
请问一下,总氮的测定中三氯甲烷的作用是作为硝酸钾储备液的保护剂,然后在保护剂的作用下,在0-6℃暗处保存,可以稳定6个月。可是目前实验室没有三氯甲烷,想购买也比较麻烦(还要去公安局备案),可以不加三氯甲烷吗?还有如果不加三氯甲烷,储备液可以稳定多久(是否需要做标准曲线,通过空白吸光度去验证?)
二氯甲烷哪家的好,分析纯,国产货就行。二氯甲烷不用备案吧。
为什么新的国标GB/T5750-2006中二氯甲烷用FID,而三氯甲烷要用ECD?二氯甲烷不能用ECD吗?不是响应的更好吗?
用什么检测器可以测三氯甲烷 什么仪器?







 我要推广仪器
我要推广仪器
 下载APP
下载APP