测定多环芳烃,用六甲基苯溶液作内标,购买的六甲基苯是固体的,请教溶液怎样配?是不是用二氯甲烷做溶剂?具体怎样操作,溶质和溶剂的量是多少?E-mail:zhoujuan@stu.xjtu.edu.cn
测定大气中多环芳烃时,利用内标六甲基苯,打算用异辛烷配制,但不知道其在异辛烷中的溶解度或者饱和度,哪问高手知道指教一下?有没有做过的,讲授一下在配备过程中的注意事项。多谢啦
《六甲基二硅胺烷》的标准,谢谢先!!!!
六甲基二硅胺烷和三甲基氯硅烷作为硅甲基化试剂与醇的反应机理! 另外,六甲基二硅胺烷可以与醇反应,为什么还要加入三甲基氯硅烷?三甲基氯硅烷不是也可以与醇反应吗?请指教!
求助:六甲基二硅醚的检测方法?[em09509] 帮帮我~!!!谢谢了哦
用气相色谱检测奶粉中的肌醇时,用的是肌醇与三甲基氯硅烷 六甲基二硅胺烷 NN二甲基酰胺等硅烷化试剂,我想请教一下大神们肌醇与这些试剂的反应产物是什么,能提供相关的反应原理或者反应式吗??
各位老师前辈们好,我的质谱仪平常是检测水、土VOC自动进样器的,需要的时候跟换进样口至热解析仪。最近发现无论是做水、土、气VOC(三个是不同的升温程序),在中间都会出现一个较大的杂峰,用超纯水空白做也是,谱库查询后是六甲基二硅氧烷的分子式。这是什么问题?请各位老师指导一下。
今天配制过程如下: 移取100uL三甲基氯矽烷(TMS)入1.8mL进样小瓶,然后再移取300uL六甲基二矽烷(HMDS)入之,一切正常,但是再移取900uL吡啶入之时,先出现“白雾”,然后再出现固态“沉淀”。 奇怪的是这沉淀漂在上面,振摇,沉淀减少,而且底部的衍生试剂开始澄清。 这正常吗?
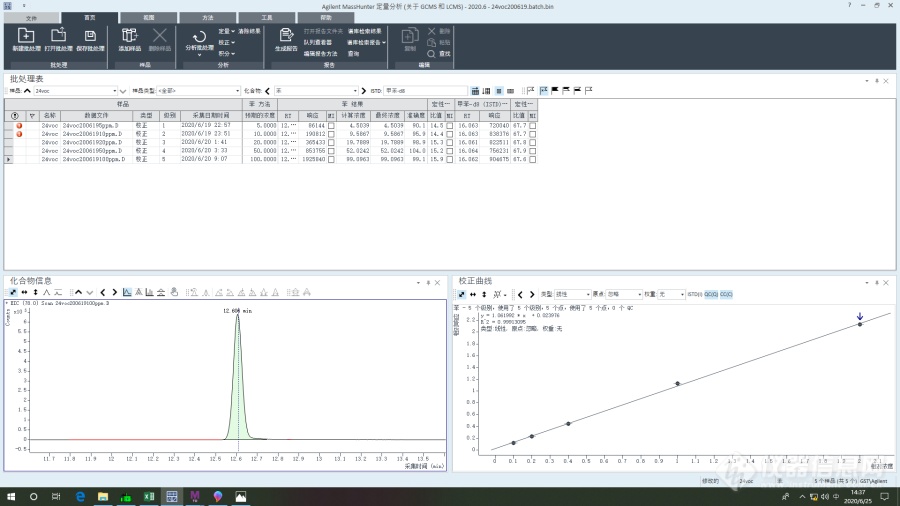
刚接手热脱附-[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url],最近在方法验证,用HJ734测废气中的24种voc,内标法。结果发现其他物质的线性、相应都比较好,唯独六甲基二硅氧烷相应很低,线性很差,基本上就不成线性。也问了身边做过734的同行业以及仪器工程师都说没有发现有这种现象,特地向大家请教有没有遇到过这种现象以及该如何解决。试过混标不经过热脱附直接液体进样,直接液体进样六甲基二硅氧烷相应和线性都比较良好,所以感觉问题还是出在[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]前面,热脱附这一块:标样的吸附加载、样品管的解吸、冷肼吸附解吸。也找过热脱附的工程师,热脱附条件也都没有问题,而且是单单六甲基二硅氧烷出现这样的问题,在紧挨着六甲基二硅氧烷之前之后出峰的乙酸乙酯、苯相应线性都很好。热脱附是Markes的TD100-xr,[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]是安捷伦8860-5977B[img=六甲基二硅氧烷曲线,690,387]https://ng1.17img.cn/bbsfiles/images/2020/06/202006251449090361_3217_2754730_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2020/06/202006251449444284_7962_2754730_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2020/06/202006251450329478_2733_2754730_3.png!w690x387.jpg[/img]三张分别为同一个系列做出来的六甲基二硅氧烷、苯、乙酸乙酯曲线,六甲基二硅氧烷基本上不成线性且相应很小[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2020/06/202006251453282683_3933_2754730_3.png!w690x387.jpg[/img]这是热脱附的条件[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2020/06/202006251454484619_8086_2754730_3.png!w690x387.jpg[/img]这是[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]这边升温条件
群友问:用GB 5413.25-2010 第二法气相色谱法做肌醇,实验室买的三甲基氯硅烷和六甲基二硅胺烷有本底干扰,不知道老师们这两个试剂买的是什么牌子的?群友一:国药的群友二:三甲基氯硅烷,扫尾剂级别,100ml,国药有的,六甲基二硅胺烷,国扫尾剂级别,25ml,也是国药的;这两个试剂SIGMA有专用的衍生剂级别的。非常感谢乳制品检测之家群友岑老师、人气老师的分享!
各位大侠: 紧急求助! 使用离子阱质谱分析六甲基硅氧烷时出峰为149,151,153,而NIST谱库谱峰为147,148,149,四级杆[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]分析结果与NIST类似,请问是什么原因?先谢了,望各位不吝赐教!
请问如何分析苯甲酸、苯二甲酸(含异构体)、苯三甲酸(含异构体)、苯四甲酸(含异构体)、苯五甲酸、苯六甲酸。用氢离子火焰型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]。
我做六甲基磷酰三胺水溶液分析,用的101硅藻土担体,涂PEG20M,水峰是拖尾的,可以选择其他哪些做担体呢? (最高使用温度250)使用的是3mm*2000mm填充柱
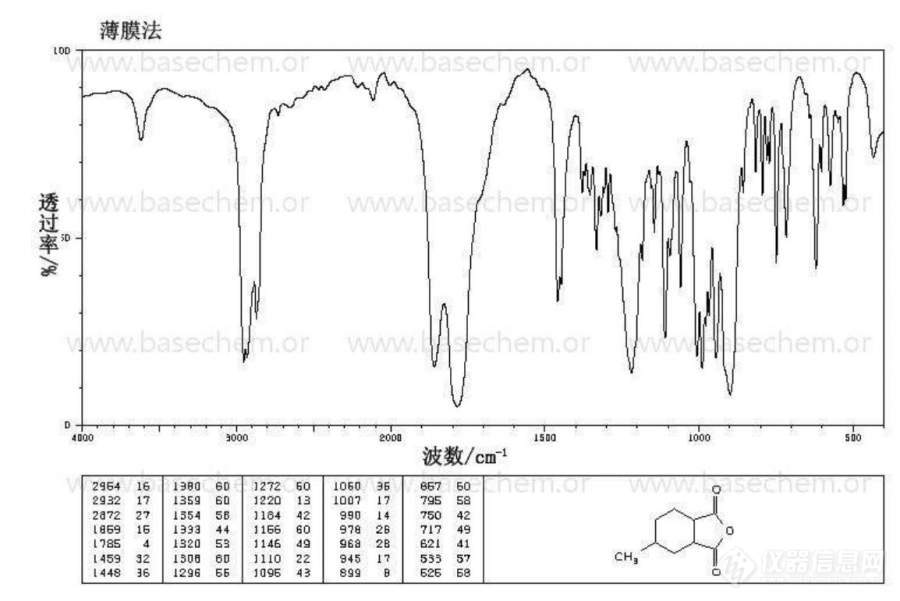
[color=#444444]我在分析甲基六氢苯酐的红外光谱过程中,无法确定900cm-1的强吸收峰的归属,希望有人能帮下我[/color][color=#444444][img=,690,460]https://ng1.17img.cn/bbsfiles/images/2019/08/201908281619247151_5896_1823055_3.jpg!w690x460.jpg[/img][/color][color=#444444][color=#008000]甲基六氢苯酐红外.JPG[/color][/color]
SVHC第八批物质种有一组甲基六氢邻苯二甲酸酐,是由4个同分异构体组成,CAS号分别为:25550-51-0 19438-60-9 48122-14-1 57110-29-9但是就其分子式来看应该只有三种同分异构体,为什么会有4个CAS号呢,或者是船式和椅式的构象不同造成的4种同分异构体?
最近在看GB5009.19-2008的方法,其中有一个农残 五氯苯基硫醚 从网上找不到CAS号,只能找到甲基五氯苯基硫醚,这两种物质是一样的吗?哪位大侠知道?
甲基六氢苯酐用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]和[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测试结果,[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]结果远大于[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]结果,那位大神能详细解答?
硫化氢的对氨基二甲基苯胺物质冻结成块了,这该怎么配?
[color=#444444]2,4-二甲基苯硫酚,2,6-二甲基苯硫酚位置异构体。沸点分别为207-208 °C,76℃[/color][color=#444444]使用Scientific TRACE TR-5MS 色谱柱,两个峰出在一起,相差0.1min,无法基线分离。尝试多种升温程序,只有保留时间和峰宽有变化,分离情况不变。降低载气流速也一样。[/color][color=#444444]换用 安捷伦vf-WAXms柱,彻底分不开了。瘦高的一个峰。[/color][color=#444444]两个柱子都试过60,70,90,120度恒温,分不开。梯度升温,也分不开。[/color][color=#444444]请各位大侠帮帮忙,试了两天了。[/color]
我用气相色谱分析脂肪醇,用吡啶溶解,再加三甲基硅烷时,出沉淀,不分层!继续加六甲基二硅胺烷时,出现分层,下面是沉淀,请问沉淀是什么物质?请问有知道的吗?急!
哪位同道有 对氨基二甲基苯胺比色法 GB 18056 –2000 附录A 用来检测甲硫醇的,急需!!!
需几个标准,哪位大侠有,帮偶找一下HG/T 3925-2007 甲基苯骈三氮唑 HG/T 2519-2007 工业六聚偏磷酸钠 HG/T 937-2007 工业用1,6己二胺 HG/T 3923-2007 循环冷却水用再生水水质标准 另还有四乙烯五胺的标准,多乙烯多胺非常感谢!
急求文献一篇!非常感谢!!!碘量法测定邻甲基苯硫脲 选自《理化检验-化学分册》2003 年3 月39 卷3 期 中文版
樟脑苯扎铵甲基硫酸盐原料炽灼残渣后残渣还有25%左右,但是原料标识含量为97%,请问,如何去确定残渣的含量?
在合成六甲基四氢萘中(以对异丙基甲苯和2,3-二甲基-1-丁烯在环己烷做溶剂,叔丁基氯做吸氢剂,无水氯化铝做催化剂的情况下,合成得到1,1,3,4,4,6-六甲基四氢化萘(HMT),先想用气相色谱确定反应生成了多少HMT,怎么办?实验中选择对二甲苯做内标,测定HMT对二甲苯的校正因子,但是所测得校正因子不稳定。希望大家多多帮助!
我现在用agilent 6820 作原料药中的 - 甲苯和二甲基甲酰胺(DMF)残留 。用甲醇多样品进行溶解, 我现在摸索到的条件: 35度 保持1 分钟; 5度/分钟 到70 度 ,甲醇的出峰时间是1分钟之前, 甲苯2分钟多点 ,DMF 在3 分左右 , 感觉不妥之处是:即使最后的70 度,也远低于甲醇,甲苯、DMF 的沸点 ? 是不是做错了阿? 请大侠指点
如题,测试六甲基环三硅氧烷(d3)——保留时间为5.06min-6min,八甲基环四硅氧烷(d4)——保留时间为8.38min,十甲基环五硅氧烷(d5)——保留时间为10.58min和十二甲基环六硅氧烷(d6)——保留时间为13.26min时,发现前面两种物质(d3和d4)时,峰很怪异,d3的峰是个土包,d4的峰前面突出一块,而d5和d6的峰非常好!我改变了进样口温度,离子源温度,初温等,均未改善!我使用的GC/MS条件如下:进样口温度140℃,离子源和传输线温度为160℃,升温程序:40℃(保持3分钟),以10℃/min升至150℃,再以30℃/min升至300℃(保持4分钟)。我降低进样口温度至120℃时情况改善一点,但是d5和d6没法汽化了,昨天还截了柱头,换了衬管、隔垫均无济于事!请各位大虾帮忙解决,谢谢!附件为scan扫描的谱图。
对氨基二甲基苯胺光度法测定水样中硫化物的几个问题,希望那个大侠帮忙解决一下?我们公司使用的工艺是低温甲醇洗,测定的是其贫甲醇中的微量硫化氢含量?在测定过程中遇到的问题是:1、回收率达不到95%以上;2、用不同的稀释倍数测定同一个样品,其测定结果相差很大。3、贫甲醇中的硫化氢含量其实不多,但是把上述方法中药品加进去后,其颜色一直很深,且最后不会变成蓝色,但是用硝酸银定性,又感觉硫化氢很多。
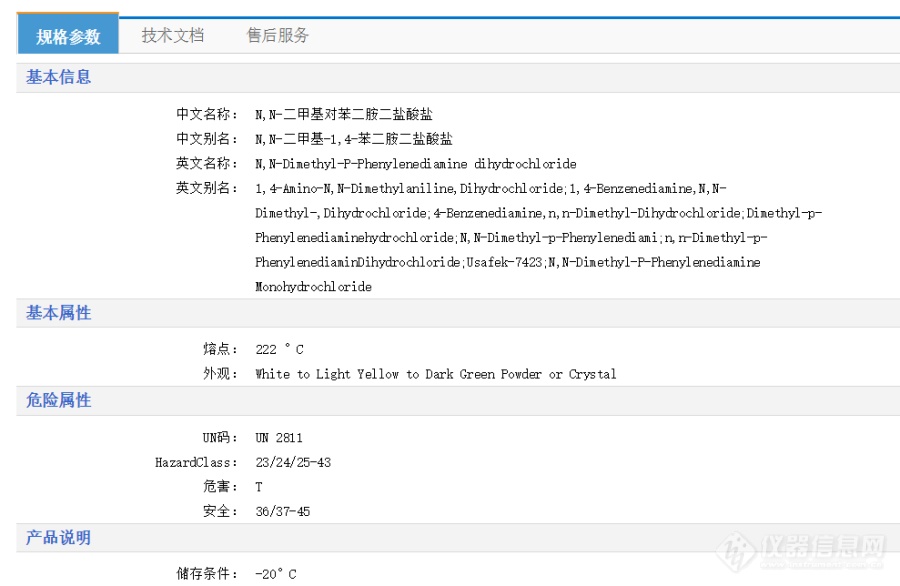
1. 在用亚甲蓝测水中硫化物的时候,需要配置0.2%的对氨基二甲基苯胺溶液,我查了一下,他的别名也可以叫叫[font=宋体, Arial, Helvetica, sans-serif][size=12px]N,N-二甲基对苯二胺二盐酸盐,应该没买错吧。[/size][/font]2. 他的储存条件不是-20℃吗?这样它里面会不会含有很高的水分?那我称取2 g的 时候,需不需要干燥,还是直接就称取2 g 就好了。3. 还有就是配置好0.2%的对氨基二甲基苯胺溶液后,怎么储存呢?室温?冷藏?冷冻?如有回复,万分感谢!![img=,690,449]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239193272_1018_5383916_3.png!w690x449.jpg[/img][img=,690,299]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239240158_2694_5383916_3.png!w690x299.jpg[/img]
年初六家里被盗,两个本本都没了,连移动硬盘和u盘都拿走了。polic笔录的时候问起来,我竟然连自己电脑的型号之类的相关信息都不知道。太菜了。准备买个新的,大家建议下,我该怎么选择,学习的话从那里开始学啊







 我要推广仪器
我要推广仪器
 下载APP
下载APP