根据GB/T 《小麦粉与大米粉及其制品中甲醛次硫酸氢钠含量的测定》做空白试剂加标。本标准的检出限0.08ug/g。我做得标准曲线的浓度是0.002、0.005、0.01、0.02、0.05ug/ml。空白试剂加标水平为1倍检出限:1.6ug、2倍检出限:3.2ug、10倍检出限:16ug。实验过程:1、(本标准是称取20.0g样品置于250ml具塞三角瓶中,加入200ml盐酸-氯化钠溶液)本人做的是空白试剂加标,直接将标准品加入250ml具塞三角瓶中,加入200ml盐酸-氯化钠溶液,摇匀,备用。2、取上述溶液2.0ml于25ml比色管中,加入1ml磷酸氢二钠溶液、0.5ml衍生剂,补加水至10ml,盖上盖子,摇匀。置于50℃水浴锅中加热40min后,取出用流水冷却至室温。3、准确加入5.0ml正己烷,将比色管横置,水平方向轻轻振摇3~5次后,将比色管倾斜放置,增加正己烷与水溶液的接触面积。在一小时内,每隔5min轻轻振摇3~5次,然后再静置30min,高效液相色谱法测定。讨论:为什么进针出来的峰形很难看,标准曲线也特别差,而且每次跟每次进针峰都会变,跟标准品甲醛不稳定有关系吗?各个加标水平回收率也特别不稳定。急切希望高人指点一二!
今天做氨氮的时候发现酒石酸甲钠结晶了!大家知道是啥原因啊?又办法解决啊?麻烦大家了!
做氨氮时 加酒石酸钾钠水变浑浊 ~~~ 很头疼 换了酒石酸钾钠还是不行 可能是水样的问题 看反应原理 是用来屏蔽钙镁离子干扰的 能不能用EDTA-2Na来代替 那位DX知道的 来说说~~~行不行~~
领导要求做碳酸氢钠的X衍射,想知道碳酸氢钠的晶型,但没有相关的操作规范和标准,哪位前辈有相关资料,请指点下,在线等....
用戴安的离子色谱,最近想分离溴酸根和氯离子,淋洗液是碳酸钠和碳酸氢钠,试了不同的配比都无法分开,想单独用碳酸氢钠做淋洗液不知道可行吗?
做COD标样后剩下的标样下次再用,如何保存?放入冰箱浓度会变吗?我不太敢往里加硫酸,因为是标样,比较珍贵吧.
http://simg.instrument.com.cn/bbs/images/brow/emyc1007.gif这是我们实验室几年前做的,拿来参加原创大赛,支持化学药分析版。HPLC法测定小儿氨酚黄那敏片马来酸氯苯那敏含量【处方】 马来酸氯苯那敏 0.5g 对乙酰氨基酚 125g 人工牛黄 5g共制成 1000片1.对照品与供试品马来酸氯苯那敏对照品(中国药品生物制品检定所,批号100047-200305)对乙酰氨基酚对照品(中国药品生物制品检定所,批号100018-200408)小儿氨酚黄那敏片(本公司,批号:20060201、20060801、20060802)小儿氨酚黄那敏片阴性样品(不含马来酸氯苯那敏和对乙酰氨基酚、本公司)2.马来酸氯苯那敏含量测定2.1仪器与试剂2.1.1仪器:岛津LC-10A高效液相色谱仪2.1.2试剂:甲醇、乙腈为色谱纯,磷酸二氢钾、三乙胺、磷酸为分析纯,水为超纯水。2.2测定方法【含量测定】照高效液相色谱法(中国药典2005年版二部附录V D)测定色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;磷酸盐缓冲液(分别取乙腈250ml与0.02mol/L磷酸二氢钾溶液250ml加十二烷基磺酸钠0.68g,振摇使溶解,用磷酸调pH至3.5)-甲醇(10:9)作为流动相,检测波长为215nm,理论塔板数按马来酸氯苯那敏峰计算应不低于3000,马来酸氯苯那敏峰与其他峰的分离度应符合规定。对照品溶液的制备 取马来酸氯苯那敏对照品约20mg,精密称定,置50ml容量瓶中,加甲醇-0.5%醋酸(1:1)混合液适量,振摇使溶解,加上述混合液稀释至刻度,摇匀,精密量取5ml[/fon
哪有做氨基酸的强酸性阳离子树脂 钠型(分析用)卖? 做氨基酸的强酸性阳离子树脂 钠型(分析用)卖? 分离和反应用的 仪器是日立的,柱子好象没国产的
做农残实验时,提取水稻植株中的二氯喹啉酸时加二氯甲烷?为什么还要加饱和碳酸氢钠溶液?碳酸氢钠溶液有什么作用?
污水厂出水加入酒石酸钾钠后出现浑浊,吸光值增大,可以不加酒石酸钾钠吗,做曲线的时候加酒石酸钾钠不会出现浑浊的现象
做苯甲酸,山梨酸和糖精钠很长时间了,一直分离的很好,峰形也很好,但最近一个月,几个峰开始拖尾严重,以致到现在山梨酸的保留时间延长和糖精钠根本分不开了,请问大家有碰到这种现象吗?我用的流动相是甲醇:乙酸铵=5.5:94.5。我最近换了针筒过滤器,它会带来这样的影响吗?请大家多给宝贵意见。谢谢!
玻璃要泡酸,众所周知,那塑料的容量瓶要泡酸吗?为什么呢?最近测食品钠钙老是不稳定。
用TENAX采样管进样做TVOC,为什么乙酸正丁酯出不来?求解?
请问专家我们严格按照国标的方法同时做苯甲酸,山梨酸和糖精钠,可发现出峰顺序与标准不一样,标准是苯甲酸山梨酸,糖精钠,可我们是苯甲酸,糖精钠,山梨酸,怎么会这样?
今天用液相色谱仪检测依地酸钙钠中的氨基三乙酸,用的是安捷伦的C8柱子,流动相为0.01mol/L的氢氧化四丁基铵:甲醇=90:10,溶剂为硝酸铜,可是压力很不稳定,好不容易平衡后,一进样,压力瞬间飘很高,在走样过程中逐渐下降,走一针过后,基线很不稳定,感觉柱子里面冲出来很多东西,有没有做过这个品种的前辈们啊,知道我一下吧,怎么改进?
用TENAX采样管进样做TVOC,为什么乙酸正丁酯出不来?求解?
食品中硝酸盐亚硝酸盐测定能用碳酸钠碳酸氢钠系统做吗?新国标中是用AS11-HC,KOH梯度做我的机子,太老了,DX120的,碳酸钠碳酸氢钠系统,没有梯度。AS9等度能做吗?或者碳酸钠碳酸氢钠系统有什么柱子合适呢?求助啊。。。[em09509]

按国标方法做苯甲酸山梨酸糖精钠,流动相是甲醇:乙酸铵http://ng1.17img.cn/bbsfiles/images/2016/06/201606070935_596198_2775594_3.jpg5:95,5分钟不到峰全部出了,分离也不好,不知道是什么原因,求大神分析下可能原因
氘代三氟乙酸做溶剂核磁共振氟谱出峰位移是多少啊?怎么查化合物氟谱位移啊,有什么书总结吗?
实验室经常做的项目如果申请CNAS认可的话算扩项吗?
我的铬标准溶液分别为20ppb和10ppb的2%(V/v)硝酸溶液(这个酸浓度的确有点高),清洗进样针的硝酸也为2%。但是这根进样针用的比较久了,同一个浓度的进样,信号值会越来越高。石墨炉的清洗是没有问题的,已经排除,已经锁定进样针清洗不干净。如果不解决这个问题,做完10ppb的质控点以后的那个样品,基本上要重新做。这样不仅仅浪费时间,还费石墨管的使用寿命。机器是PE的AA800请教方家,我是不是该把铬标准溶液的酸浓度调低到0.3%左右?这样子清洗进样针的硝酸保持2%的浓度,可以吗?大家平时的标准溶液和清洗酸浓度是怎么样的?如果进样针内有点长菌该怎么清洗?
依地酸二钠BP2015中有关物质所用色谱柱填充料为:— stationary phase: spherical graphitised carbon for chromatography R1 (5 μm) with a specific surface area of 120 m2/g and a pore size of 25 nm大家用的哪个牌子的柱子?麻烦告知一下,谢谢!
我使用Agilent 的ZORBOX SB C18柱子做苯甲酸山梨酸糖精钠,刚开始很好,做了几小时后峰形就变了时间也提前很多。柱子原来使用过,做苏丹红用过,我以为是用久了的原因,就换了一根新的SB C18柱子,情况也一样。我就搞不懂了,不就是用0.02mol/l的乙酸铵和甲醇做流动相嘛,怎么会这样?做苏丹红的时候连续使用了几周没停过,重现性都好得很。我用迪马的柱子做苯甲酸山梨酸糖精钠,都没出现这种情况。难道SB C18柱子不能使用乙酸铵做流动相?是不是固定相被损坏或者起了反应?能不能再生?怎么再生?可不可以用三氯甲烷?
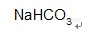
【中文名称】碳酸氢钠;酸式碳酸钠;重碳酸钠;小苏打;焙烧苏打;重碱【英文名称】sodium bicarbonate;sodium hydrogen carbonate【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/03/201203072013_353130_1855403_3.jpg【相对分子量或原子量】84.01【密度】2.20【毒性LD50(mg/kg)】 大鼠经口4300【性状】 白色单斜晶体。【用途】 是重要的常用药物(消化剂、制酸剂),又是制灭火剂、焙粉和清凉饮料等的原料。还可用于饲料作饲料添加剂的缓冲剂。【制备或来源】 可由碳酸钠浓溶液或结晶碳酸钠吸收二氧化碳而制的。是氨碱法制纯碱的中间产物。【其他】 在热空气中,能缓缓失去一部分二氧化碳,加热至270℃失去全部二氧化碳。
请问做食品中山梨酸、苯甲酸和糖精钠的朋友,我前天刚开始做这三种物质的检测,走的标准样。如果按照国标流动相用甲醇:乙酸铵=5:95比列,流速1.0ml/min进标准混合样,柱温是40°。进样结果30分钟内无峰。如果把流动相改为甲醇:乙酸铵=50:50比列,则可以分出三个峰,但是国标上第三个峰是糖精钠,而我做的第一个峰是糖精钠。哪位大侠帮忙分析一下这是什么原因呢?我用的仪器是岛津LC-20AT。
以前一直在用一直没事 昨天消解完 发现主罐喷出绿色液体 应该是 管子里有水被探测针烧穿 导致酸回流喷出还腐蚀了探测针请问换新的后 我里面不放酸只放纯水可行吗 主罐主要是为了放探测针测温度的吧 换成水会影响设置好的温度吗
做氨氮,新买的酒石酸钾钠分析純,配置“酒石酸钾钠”500g/l溶液呈现黄色,以前配置的时候都是无色的,求解怎么回事
[color=#444444]各位高人:有谁做过[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]分离依地酸二钠和氯离子?方法是什么指导一下吧,我做的依地酸二钠总是峰型拖尾严重。[/color][img=,absmiddle]http://muchongimg.xmcimg.com/data/emuch_bbs_images/smilies/tongue.gif[/img]
有哪位前辈做过马来酸氯苯那敏的残留溶剂,想请教一下,前辈们做的色谱条件和方法,我按照2015版药典做的,吡啶峰出不来
做氨氮的纳氏试剂分光光度法的时候,样品加了酒石酸钾钠后,产生了很多白色悬浮物,这个时候样品应该做什么前处理呀?







 我要推广仪器
我要推广仪器
 下载APP
下载APP