各位大佬,现在实验室用BJS202002做苯甲羟肟酸,但峰型经常拖尾,时好时坏。换柱子,换设备都试过,但也找不到原因,各位大佬有没有遇到类似情况?仪器用过LC-20和U3000,柱子用过艾杰尔、安捷伦、依利特的,都是不稳定,今天峰型好了,明天又拖尾[img]https://simg.instrument.com.cn/bbs/images/default/em09504.gif[/img]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=127755]对羟甲基苯氧乙酸的合成[/url]
《ChP2005》二部中,羟苯乙酯的鉴别:取本品约0.1g,加乙醇2ml使溶解,煮沸,加硝酸汞试液0.5ml,放置后逐渐生成沉淀,上清液显红色。开始是发生水解反应,可是水解产物与醋酸汞反应原理是什么?
瑞士万通的905自动电位滴定仪,搭配光度电极。用EDTA滴定镍钴锰溶液的总量。如果滴定溶液中不加盐酸羟胺,初始电位很低很低,突越很不明显,加入盐酸羟胺初始电位就有700多mv,正常。但是观察溶液颜色,加入盐酸羟胺前后基本无变化。这是什么原因?滴定用的是可见光波长。
在检测水产品中孔雀石绿残留量的标准方法中,很多都要用到二甘醇、盐酸羟胺及对-甲苯磺酸,但不知它们的作用是什么,不知哪位知道,大家都来谈一谈啊

作者:http://d.g.wanfangdata.com.cn/Images/head_pic.gif张琰 http://d.g.wanfangdata.com.cn/Images/head_pic.gif陈文秋 http://d.g.wanfangdata.com.cn/Images/head_pic.gif余敏灵 Author:Zhang Yan Chen Wenqiu Yu Minling 作者单位:四川省眉山市人民医院,四川,眉山,620010 四川省乐山食品药品检验所,四川,乐山,614000摘要: 目的 建立测定羟苯乙酯中水杨酸含量的方法 .方法 采用反相高效液相色谱法,Diamonsil C18柱(250mm×4.6 mm,5μm),以0.05mol/L磷酸二氢钠缓冲液(pH=3.4)-乙腈(50:50)为流动相,检测波长为297 mm.结果水杨酸质量浓度在0.51~13.26 μg/mL范围内与峰面积呈良好的线性关系,平均回收率为99.0%,RSD=0.8%(n=6).结论 该法简便、准确、快速,适用于羟苯乙酯中水杨酸的含量测定.http://ng1.17img.cn/bbsfiles/images/2012/08/201208201739_384784_2379123_3.jpg
各位大大,小弟最近在做醇酸树脂酸值和羟值的滴定,羟值滴定的方法是用乙酸酐--4-DPMA的方法,酸值测定结果为21mg KOH/g 样品。羟值直接滴定的结果为-14mgKOH/g 样品,出现负值是不是因为羧基与滴定剂氢氧化钾反应造成的?根据方法羟值=酸值+测定的羟值,也就是21+(-14)=7mgKOH/g 样品。那这7mgKOH/g 样品就是样品羟值吗?照这样计算样品羧基中的羟基好像没有包含在结果中,现在感觉很纠结。
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.
试问:硫酸铈能否氧化对叔丁基苯酚?可以直接电位滴定测对叔丁基苯酚的纯度吗?实验室电位滴定仪:梅特勒T50。若手动滴定,指示剂采用二苯胺磺酸钠还是邻二氮杂菲-亚铁?望指点
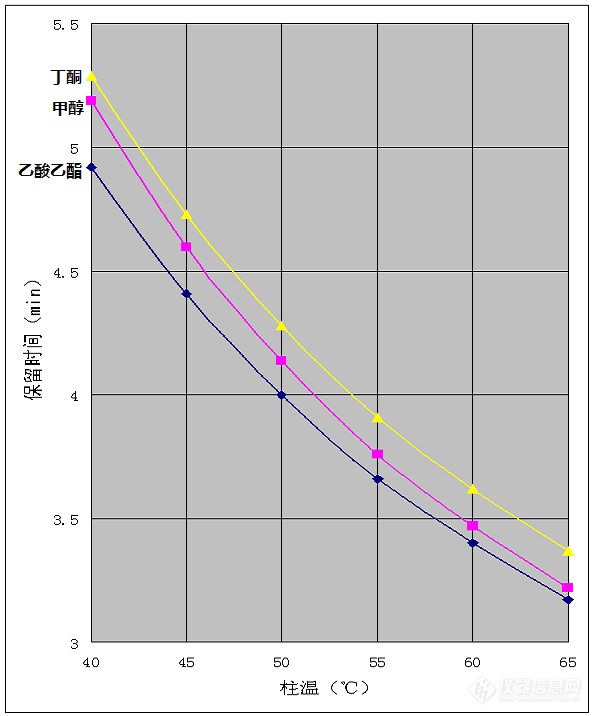
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。
如果要在固定污染源(排气筒)中采集甲苯、乙酸乙酯这样的有机物,通常情况是不用加热枪杆采样的,那么当遇到水汽成分(也就是含湿量)很大的时候,要不要用加热枪杆呢?做过这个方面采样的版友,是用什么仪器采样的?你们的仪器有没有配备干燥器?我们的仪器只有配加热枪杆,现在就不用加热枪杆的话,所以连接方式就是“排气筒→采样皮管→活性炭管→采样皮管→采样仪器”。
我用乙酸乙脂做溶剂,溶解丙二酸,丁二酸,苯酚等物质,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测,只有溶剂峰出现,其他都没有信号,采用程序升温从120度到180度检测了60分钟左右,求助各位高手为什么其他都没有出峰呢?
刚做的对氯苯氧异丁酸怎么进行含量测定?要是用HPLC条件怎么得到? 如题,谢谢各位朋友。
如题有二甲苯 醋酸丁酯 醋酸戊酯 芳烃 醋酸异丁酯 用什么分离条件好呢?
一直糊里糊涂的做,多方查证后得知,与大家分享:盐酸羟胺:作为还原剂,有助于防止待测物的降解;对甲苯磺酸:作为离子对试剂,有利于孔雀石绿形成离子对,增加乙腈对该物 质的提取效率;氧化铝:吸附样品中的油脂;二甘醇:消除乳化。
求助三氯吡氧乙酸、2,4-二氯苯氧乙酸正丁基酯(2,4-D丁酯)的检测方法

今天在整理记录,翻开药典,发现“甲乙丙丁”都有收载。羟苯乙酯 化学名:对羟基苯甲酸乙酯 别名:尼泊金乙酯 本品是用对羟基苯甲酸与乙醇酯化而得。是属于酯类,常作抑菌防腐剂。中国药典2010年版二部正文品种第二部分收载有羟苯乙酯,具体页数为1223页http://ng1.17img.cn/bbsfiles/images/2014/09/201409250848_515487_1621890_3.png其检测方法均相同:色谱柱信息:welchrom c18 pn:00310-02041,sn:w132111841 4.6*150mm取本品加流动相溶解并稀释制成每lml中含1mg的溶液,作为供试品溶液;精密量取lml,置100ml的量瓶中,加流动相稀释至刻度,摇匀,作为对照溶液。照高效液相色谱法(附录V D )试验,用十八垸基硅垸键合硅胶为填充剂,以甲醇-1%冰醋酸溶液(60:4 0 )为流动相,检测波长为254nm。取羟苯甲酯和羟苯乙酯,加流动相溶解并稀释制成每1ml各含10μg的溶液,取20μl注入液相色谱仪,记录色谱图,羟苯甲酯峰与羟苯乙酯峰之间的分离度应符合要求。取对照溶液,注人液相色谱仪, 调节检测灵敏度, 使主成分色谱峰的峰高约为满量程的25% ; 再精密量取供试品溶液与对照溶液各20μl,分别注人液相色谱仪,记录色谱图至主峰保留时间的4 倍。供试品溶液色谱图中如显杂质峰, 单个杂质峰面积不得大于对照溶液主峰面积的0.4倍(0.4% ) , 各杂质峰面积的和不得大于对照溶液主峰面积的0.8倍(0.8% )。典型色谱图:1.系统:甲乙已经分离的很不错。http://ng1.17img.cn/bbsfiles/images/2014/09/201409250851_515488_1621890_3.png2.空白溶剂:http://ng1.17img.cn/bbsfiles/images/2014/09/201409250853_515489_1621890_3.png3.对照溶液:1%供试液的色谱峰还是刚刚的。http://ng1.17img.cn/bbsfiles/images/2014/09/201409250853_515490_1621890_3.png4.供试液:3.244min的色谱峰就是甲、6.973min的色谱峰就是丙、4.456min的色谱峰就是乙。http://ng1.17img.cn/bbsfiles/images/2014/09/201409250854_515491_1621890_3.png5.色谱图对比:一目了然。http://ng1.17img.cn/bbsfiles/images/2014/09/201409250855_515492_1621890_3.png6.本次试验主要是没有羟基丁酯,这个是个辅料,下次检验,借点羟基丁酯,让路人甲乙丙丁汇聚一堂。以上试验结果表明月旭色谱柱性能优良。
请教各位大神:甲苯和醋酸仲丁酯混合后经检测为何只出一个峰值? 甲苯和醋酸仲丁酯单独进样出峰时间都是一样的!后将载气流速调慢,结果是一样的!请问怎样才能将这两种物质区分开来?
比硫酸強的上千倍的酸-超強酸超強酸(英文:Superacid)是指比純硫酸酸性更強的酸。簡單的超強酸包括三氟甲磺酸(CF3SO3H)和氟硫酸(FSO3H),它們的酸性都是硫酸的上千倍。在更多的情況下,超強酸不是單一純淨物而是幾種化合物的混合物。超強酸這一術語由詹姆斯布萊恩特科南特(en:James Bryant Conant)於1927年提出,用於表示比通常的無機酸更強的酸。喬治安德魯歐拉因其在碳正離子和超強酸方面的研究獲得1994年諾貝爾化學獎。魔酸(Magic acid)是最早發現的超強酸,稱它有魔法是因為它能夠分解蠟燭中的蠟。魔酸是一種路易斯酸五氟化銻(SbF5)和一種質子酸氟硫酸(FSO3H)的混合物。目前已知最強的超強酸是氟銻酸(en:Fluoroantimonic acid),一種氫氟酸(HF)與五氟化銻(SbF5)的混合物。其中,氫氟酸提供質子(H+)和共軛鹼氟離子(F− ),氟離子通過強配位鍵與親氟的五氟化銻生成具有八面體穩定結構的六氟化銻陰離子(SbF6− ),而該離子是一種非常弱的親核試劑和非常弱的鹼。於是質子就成為了「自由質子」,從而導致整合體系具有極強的酸性。氟銻酸的酸性通常是純硫酸的2×1019倍。2004年,加州大學河濱分校的Christopher Reed研究小組合成出了一種號稱史上最強的純酸—碳硼烷酸。由於碳硼烷酸中碳硼烷的結構十分穩定且體積較大,一價負電荷被分散在碳硼烷陰離子的表面,因而與氫陽離子的作用很弱,碳硼烷酸從而具有令人吃驚的釋放氫離子的能力。據他們報道碳硼烷酸的酸性是氟硫酸的一千倍,純硫酸的一百萬倍,水的100萬億倍;而釋放氫離子後,碳硼烷的結構(由11個硼原子和一個碳原子排列而成的20面體)不會輕易發生變化,難以進一步發生化學反應,因此腐蝕性很低。這種新的既超強又溫柔的固體碳硼烷酸可以質子化很多物質,如C60。而Reed企圖用它酸化惰性氣體(應該叫稀有氣體),來看看這些氣體倒底有多懶惰。碳硼烷酸在催化和制藥領域也有很廣闊的應用前景。不過,美中不足的是,碳硼烷酸的產率很低,還只局限於世界上少數幾個實驗室的研究。
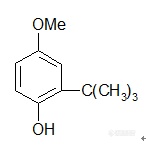
【中文名称】3-叔丁基-4-羟基茴香醚;3-叔丁基-4-羟基苯甲醚;2-叔丁基-4-甲氧基苯酚【英文名称】3-t-butyl-4-hydroxy anisole【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/03/201203241952_357143_1855403_3.jpg【相对分子量或原子量】180.25【熔点(℃)】48~55【沸点(℃)】264~270℃(9.77E4Pa)【毒性LD50(mg/kg)】 混合物大鼠经口2200【性状】 与3-叔丁基-4-甲氧基苯酚混合为无色至微黄色蜡状固体或粉末,略有刺激性气味。【溶解情况】 不溶于水。【用途】 市售品为3-叔丁基-4-甲氧基苯酚和2-叔丁基-4-甲氧基苯酚的混合物,用于食品用抗氧剂,抗氧能力3-叔丁基-4-甲氧基苯酚强,两者同时使用,起协同作用,可用于油脂、油炸食品、干鱼制品、饼干、方便面、罐头及腊肉制品等。除单独使用外,亦可与抗坏血酸、异抗坏血酸、柠檬酸等配合使用。【制备或来源】 (1)以对羟基苯甲醚为原料,在硫酸或磷酸催化剂存在下,在80℃下与叔丁醇反应而得混合物;(2)以对苯二酚和叔丁醇为原料,反应生成叔丁基对苯二酚,再以锌粉为催化剂,与硫酸二甲酯进行甲基化反应而得混合物。【其他】 遇铁离子会着色。与BHT、没食子酸丙酯、对苯二酚、蛋氨酸、卵磷脂、硫代二丙酸等有协同作用。【生产单位】略
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
请教各位,想对醇酮类,醋酸丁酯/乙酯,苯类混合物做气相,用什么色谱柱比较好?谢谢http://simg.instrument.com.cn/bbs/images/default/em09511.gif
含有羟基丙烯酸的乳液羟值怎样测定?因带有羰基,直接测定肯定不行,有什么方法可以保护羰基,请各位老师指点。
我们现在需要测固定污染源中的乙酸丁酯和二甲苯,我看有些标准说用热吸附采样,还有些标准说用活性炭采样,我想问一下各位大佬,到底是用哪种采样管来进行采样。同时环境空气也是跟固定污染源用一样的采样管吗?
[align=left] 意想不到盐酸羟胺的不稳定性对实验结果的影响[/align][align=center] [/align][align=left]在前面的二篇中提及了Tris的检测,经过近一个月的努力,顺利完成了Tris的方法开发和验证。今天谈谈另一个化合物的检测验证,盐酸羟胺,在Tris快完成之际,接到另一家客户的要求,检测某一药物中残留的盐酸羟胺。[/align][align=left]天下哪有这么好的事,盐酸羟胺和Tris结构类似,理论上检测方法可以完全套用,除了淋洗液和柱子不一样外,后面的补液模式可以完全照抄!分离方法可以基本按照Tris 的方式(阳离子分离,柱后补碱,安培检测)。[/align][align=left]在开始做之前,我问了对方,盐酸羟胺的稳定性,稳定。同时,我上网简单查了一下,没有提及不稳定的事,这样完全照搬Tris的检测模式,OK。[/align][align=left]在对方厂家测试人员的协助下,按照设计的方案,排好序列,连续进样80针就够了,时间大约14-16小时,为了加快速度,通宵进行,由于担心仪器晚上会突然断开(ICS5000)我通宵查看系统,果然半夜里软件连接突然断了一次,重新连接后就没再断开了,在连续进样的过程中,我感觉有点不对,很低浓度下,怎么没有盐酸羟胺的峰(理论计算是有呀,能看到),峰感觉在变小,似乎不稳定,半夜里只能继续做下去了。[/align][align=left]第二天在分析大批数据后,终于感觉到不对了,盐酸羟胺会分解!所有实验结果泡汤了!赶快上网查资料,的确有明确提及盐酸羟胺不稳定,但如何稳定没说呀!查到相关分析的一篇类似文献,提及用流动相溶解,但其数据和结果似乎表明其检测限比我们高近10倍,他们做的很有问题。怎么办?样品不稳定这样实验做起来就麻烦了,羟胺,盐酸羟胺,我明白了,没有单独的羟胺的实际样品,只有盐酸羟胺,这就意味着只要盐酸的存在,不稳定的羟胺变成了稳定的盐酸羟胺,酸性条件稳定,也理解了论文用流动相溶解的意思。[/align][align=left]重新设计实验,盐酸羟胺稀释不用水稀释而是用跟淋洗液几乎一样的淋洗液浓度进行稀释,在酸性条件下,这样可以避免盐酸羟胺的分解。重新配溶液上机,在仅仅改变样品的溶解稀释方式,整个实验的重复性非常好,盐酸羟胺峰面积稳定回收率合格,而实验仅仅改变了一点!![/align][align=left]在实验中,由于做检测限,盐酸羟胺的浓度非常低,羟胺水解的效应非常明显,当浓度高时,不是很明显,这就是为什么一开始做了半天没发觉的缘故。对比这二个实验,Tris和盐酸羟胺结构类似,测试方法几乎一致,但完全照抄在实际中却遇到问题,说简单很简单,说复杂也不简单。Tris作为缓冲液肯定是稳定的,这是二者的差异。[/align][align=left]这也验证了做方法学的主要关键之一,样品的稳定性必须先验证。[/align]
请教各位大侠:氢氟酸是否是强氧化性酸?能否与聚苯脂发生反应?聚苯脂就是聚对羟基苯甲酸苯脂??急用,在线等。我查了很多资料都没有相关的结论。
2012年11月1号,国家安监总局对实验室间比对工作展开了,项目有活性炭管中的苯,乙酸乙酯,乙酸丁酯,我们实验室没有这些标样,购买全国都找不到货源,纠结啊。。。求助哪位仁兄帮忙介绍标样货源?标样需要活性炭管中的苯,活性炭管中的乙酸乙酯,活性炭管中的乙酸丁酯。或者二硫化碳中的苯,二硫化碳中的乙酸乙酯,二硫化碳中的乙酸丁酯。
做饮料中的山梨酸苯甲酸,根据国标用买来的山梨酸苯甲酸标准(都是1mg/ml),分别移取0.05ml、0.1、0.2、0.4、0.6苯甲酸和山梨酸到10ml容量瓶,用正己烷+乙酸乙酯(1+1)定容,配成5、10、20、40、60mg/L的浓度,但定容的过程中发现溶液好像会分层,标准曲线做出来也很不好,用丙酮做过也不行,想请问一下是什么原因,配的过程中有什么注意事项吗,配标准的手法和步骤都是一样的。
最近做了1批水样,水中可能有铁离子和铬离子。按照水质监测方法说的,加盐酸羟胺可以有效去除干扰。我想问下 盐酸羟胺是应该时候加入去除干扰,是在消解完水样后加入还是在加完1+9盐酸后加入才对?(PS:能简单解释下盐酸羟胺反应原理吗- - )
有哪些高人做过1、糠醇 2、顺丁烯二酸酐 3、甲基丙烯酸甘油酯 4、苯磺酸 5、胺菊酯、氯菊酯 。它们的GC条件是什么啊?急等回音,谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP