[color=#333333]紧急求助 22-脱氢赬桐甾醇的结构式怎么写和质谱数据。[/color]
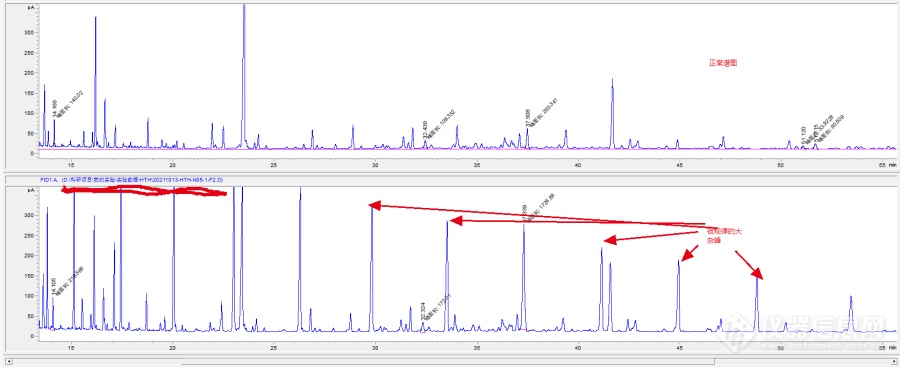
[img=[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]谱图,升温80-200-250-300-315度,检测器300°,690,281]https://ng1.17img.cn/bbsfiles/images/2021/11/202111031013369374_9410_5406430_3.png!w690x281.jpg[/img]如上图所示,萃取样品方法使用生物标志物法,空白实验无杂峰,样品实验如图中所示,有较大的杂峰,不知何原因,请各位大佬指教一二。思考如下:1.空白实验和样品实验除一个加样品,一个不加样品外,其他操作步骤均相同,样品瓶、试剂、量取仪器均同一套,如是污染的问题,但空白没污染2.这个样品之前也做过几次,均没有大杂峰出现,且是在一个样品袋里的样品,可能是样品本身的问题3.整个实验过程中,除萃取甲藻甾醇和烯酮组分,还萃取了烷烃组分,本次萃取的烷烃组分无异常。萃取顺序:先烷烃后甲藻甾醇和烯酮,如是样品问题,烷烃组分却没有异常4.此样品在仪器上测试两遍,中间间隔一周,测试结果相同。如是仪器问题,间隔一周中其他人使用该仪器无异常

10,抽取5个版友);中奖名单m3071659(注册ID:m3071659)莫名其妙(注册ID:moyueqiu)zengzhengce163(注册ID:zengzhengce163)zgx3025(注册ID:v2844608)sixingxing(注册ID:v2889187)http://ng1.17img.cn/bbsfiles/images/2016/08/201608041537_603432_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/08/201608041537_603433_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================中性甾醇方法:GC基质:标准溶液应用编号:101211化合物:5-α- 胆甾烷; 粪甾醇; 胆固醇; 菜子甾醇; 粪甾酮; 菜油甾醇、菜子甾醇; 豆甾醇; β- 谷甾醇固定相:DM-225色谱柱/前处理小柱:DM-225 30m x 0.25mm x 0.25um色谱条件:柱温:260 ℃ 恒温 载气:He, 45cm/sec, 240 ℃ 进样方式:分流, 30:1, 260 ℃ 样品:中性甾醇和植物甾醇,1.5 μL, 200ng on-column 检测:FID, 8 x 10-11 AFS, 260 ℃ 文章出处:CFR00431关键字:甾醇,食品,GC,DM-225, 5-α- 胆甾烷; 粪甾醇; 胆固醇; 菜子甾醇; 粪甾酮; 菜油甾醇、菜子甾醇; 豆甾醇; β- 谷甾醇谱图:http://www.dikma.com.cn/Public/Uploads/images/CFR00431.png图例:1. 5-α- 胆甾烷;2. 粪甾醇;3. 胆固醇;4. 菜子甾醇;5. 粪甾酮;6. 菜油甾醇、菜子甾醇;7. 豆甾醇;8. β- 谷甾醇
[color=#444444]最近在提取海洋小型浮游动物的甾醇,衍生化效果不好,导致[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测的效果不好。请问有童鞋在做这方面的研究么,想请教一二。谢谢![/color]
GDX-103色谱柱分析丙酮-异丙醇-甲基异丁基酮柱长3米,丙酮和异丙醇的峰还好,但是甲基异丁基酮的峰拖尾特别严重,调节柱温和载气流速都不怎么好使,大家能不能给点建议?
那位朋友能提供一种同时分析甾醇与甾醇的脂肪酸酯的方法,谢谢了
[color=#333333]植物甾醇能够抑制胆固醇的吸收,从而降低胆固醇。植物甾醇广泛存在于油脂和植物性食物中,例如米糠油、玉米油、芝麻油、蔬菜、水果、豆类、坚果及谷物。[/color]
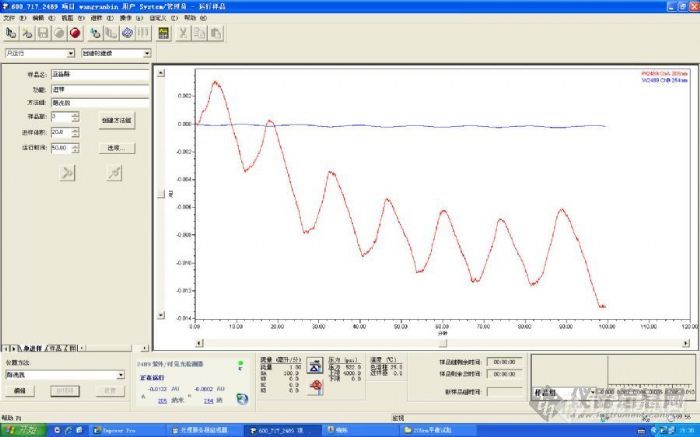
最近用205nm下检测菜油甾醇和豆甾醇,液相为waters600型,有脱气机,柱温设为25℃流动相为甲醇,默克的,在205nm下一直不能平衡,254nm是正常的。如图,请教各位老大原因。[img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903022035_136271_1833802_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903022037_136272_1833802_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903022037_136273_1833802_3.jpg[/img]
之前使用的是AJL的300SB-C18柱子,0.8ml/min,甲醇:水=90:10 ,95:5,100:0,241nm,胆甾-4-烯-3酮标品总是出现M状的峰。现在新换了一个迪马的柱子,甲醇:水=91.5:8.5,60min才出现峰,出峰时间有点过长了,但总归是觉得迪马柱子算还是不错,各位版友认为呢?有个小问题是:16min有个峰,标品也是有个小峰,不知道哪个算是胆甾-4-烯-3酮的峰?
主成分甾体皂苷除杂如色素,甾醇,脂类有什么方法啊
不知哪位是否测定过甾醇,选用什么类型的柱子较为合适啊?DB-5可以吗,谢谢!
请问如何分离乙醇、丙酮、异丙醇(三者的质量百分数差不多)。我用毛细柱,试了很多不同的载气流量和不同的柱温,进样温度,检测器温度也没能分离好,只出现一个大的平顶峰。请问有哪位前辈可以为我提供分离条件。谢谢!
[color=#444444]有个化合物确定是β-谷甾醇,但是打高分辨质谱的时候就是发不出峰,请问是怎么回事呢?[/color]
请问如何分离乙醇、丙酮、异丙醇(样品由以上三者组成,且三者的质量百分数差不多)。我用毛细柱,试了很多不同的载气流量和不同的柱温,进样温度,检测器温度也没能分离好,只出现一个大的平顶峰。请问有哪位前辈可以为我提供分离条件。谢谢!
有谁用液相检测过植物甾醇吗,我用的是外标单点校正法,但分析中发现峰面积重复性很差,每天的标样峰面积都不平行,不知是什么原因,检测条件是ODS柱,甲醇流动相,流速1.5 波长210 岛津10AVP,另外谁有完整的植物甾醇GC方法
为什么就分不开,我用的是标气其中甲醇是1500ppm,丙酮100ppm在(50度 3min,8℃/min 300)分流比5:1,氮气做载气,流速是8ml/min,FID,不知为什么出的峰就是甲醇和丙酮分不开根本就是一个峰只是甲醇略微有些拖尾,其实是丙酮,谁有跟好的条件让我借鉴一下。
请问用薄层鉴别β-谷甾醇可以用什么展开系统啊?
研究表明,当流动相的A0.7的时候,基线噪声会显著增加,一般选择的吸收值0.5。当A1.0时,基本就不能用使了。对于示差折光检测器,主要考虑样品和流动相的折光率,这里不 再赘述。选用的溶剂粘度要低、沸点适中使用低粘度溶剂,可减小容质的传质阻力,降低柱压,利于提高柱效。从分离制备和色质连用考虑,沸点要适中,低沸点的溶剂有它的优点;但仅仅用 于分析,沸点太低,反而由于溶剂的挥发,造成保留时间的变化。尽量不用高毒性的溶剂不只是环境,单从我们自身的安全考虑,当然用低毒溶剂最好。溶剂对样品有足够的溶解力样品如果不能溶解在选用的溶剂里,还怎么分析?但如果样品在流动相里溶剂仍然不大,可以用样品的最佳溶剂先溶解,再用流动相稀释,就 可以了。知道了上面选择溶剂的一般原则,就不难理解为什么不同乙醇、丙醇做流动相的原因了,因 为它们的粘度大。 还有丙酮,虽然粘度和毒性较低、溶解度大、极性适中,但用于它的截至吸收波长为330nm, 所以不常用,如果样品的吸收大于330nm,其实用丙酮是一个不错的选择。
分离甾醇用什么色谱柱比较好?什么牌子的?
请问哪位知道过氧化麦角甾醇的用途以及发展前景,拜托,拜托!
请问如何分离乙醇、丙酮、异丙醇(样品由以上三者组成,且三者的质量百分数差不多)。我用毛细柱,试了很多不同的载气流量和不同的柱温,进样温度,检测器温度也没能分离好,只出现一个大的平顶峰。请问有哪位前辈可以为我提供分离条件。谢谢!我用的是Elite 5MS的柱子。一定要用PEG20M(Carbowax20M)毛细柱才可分离吗?
各位专业的朋友们,甾醇类化合物是不是在ESI源条件下相应不好啊?并且这类物质在甲醇中溶解度不好,用二氯做的溶剂ESI源下无任何信号~是不是改换用APCI源,或是换溶剂?我是新手,望大家不吝赐教!先谢谢各位了!!
使用的是岛津2014C内标法测植物甾醇含量,和同事用的同一个内标物和对照,条件柱子都一样,为什么测出来的最后含量我的结果高了百分之九呢?([url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]小白请求各位老师指导)
各位老师: 你们好,我用茚三酮显色剂法做赖氨酸的含量,但是做的过程中发现茚三酮有点粉色,查了资料说是氧化了,所以我进行纯化,但是在纯化的过程中我发现了问题,加入热水茚三酮不溶解,还有就是加热溶解后,加入活性炭就马上析出结晶,要是这样的话,我过滤后的清液,能析出的结晶是不是就太少了? 纯化方法:称取10g活性炭,用40mL热水溶解,加入1g活性炭,搅拌1min,静置30min,过滤,将滤液放置冰箱内过夜,出现蓝色结晶,在过滤,用2mL冷水冲洗,然后再干燥器中干燥,备用。 问题1:为什么加入活性炭会出现结晶现象,良好的茚三酮和氧化的茚三酮都是同一个现象么? 问题2:干燥时是放入烘箱中进行干燥么? 问题3:有其余的纯化的方法么?麻烦老师指点一下,谢谢啦!
国家卫生部推荐植物甾醇酯的食用量为≤3.9g/d,想咨询一下大家这个数字是怎么来的,有文献或者依据可查么,谢谢!
各位大大们:有谁知道麻油里面怎么测β-谷甾醇的含量啊,要HPLC的方法,目前公司正在准备进行这方面的测定工作,可是我一点头绪也没有。江湖救急,感谢大家了
常见乙腈和甲醇作流动相进行甾醇含量分析,分享一个利用乙醇和水作流动相(绿色溶剂)[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]结合蒸发光散射检测器测定虫草药材的游离和总甾醇含量的方法。色谱柱为Poroshell 120 EC-C18 (100 mm × 4.6 mm, 2.7 μm),绿色流动相乙醇-水(89: 11),流速:0.5 mL/min,该方法可有效鉴别冬虫夏草和其他3种虫草产品,为虫草类产品质量评价提升提供了依据。详见范卫锋等,菌物学报2022.
液质联用检测血清中的植物甾醇,血样如何处理?
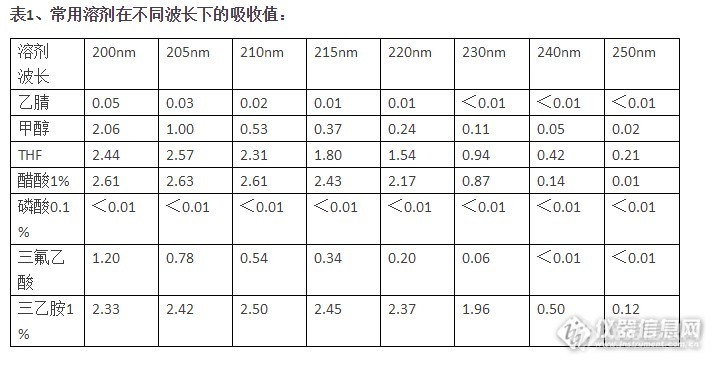
研究表明,当流动相的A0.7的时候,基线噪声会显著增加,一般选择的吸收值0.5。当A1.0时,基本就不能用使了。对于示差折光检测器,主要考虑样品和流动相的折光率,这里不 再赘述。选用的溶剂粘度要低、沸点适中使用低粘度溶剂,可减小容质的传质阻力,降低柱压,利于提高柱效。从分离制备和色质连用考虑,沸点要适中,低沸点的溶剂有它的优点;但仅仅用 于分析,沸点太低,反而由于溶剂的挥发,造成保留时间的变化。尽量不用高毒性的溶剂不只是环境,单从我们自身的安全考虑,当然用低毒溶剂最好。溶剂对样品有足够的溶解力样品如果不能溶解在选用的溶剂里,还怎么分析?但如果样品在流动相里溶剂仍然不大,可以用样品的最佳溶剂先溶解,再用流动相稀释,就 可以了。知道了上面选择溶剂的一般原则,就不难理解为什么不同乙醇、丙醇做流动相的原因了,因 为它们的粘度大。 还有丙酮,虽然粘度和毒性较低、溶解度大、极性适中,但用于它的截至吸收波长为330nm, 所以不常用,如果样品的吸收大于330nm,其实用丙酮是一个不错的选择。http://ng1.17img.cn/bbsfiles/images/2015/12/201512011315_575751_1858651_3.png
各位老师好:请问,老师有检测过药物中异丙叉丙酮和二丙酮醇痕量残留的分析的经验么?请指教。谢谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP