有谁知道5-甲基-3-庚酮的现场和共存物\干扰物?谢谢
要从甲苯 正庚烷中分离提取甲基环己烷,可以按以下步骤进行:由于正庚烷是直链烷烃,沸点比那2个相同C原子数的烃类要高,所以先蒸馏蒸出甲苯和环己烷,然后在溶液中加入酸性KMnO4,甲苯变苯甲酸下沉,分液除去上层液体,然后再用乙醚萃取几次,再酸洗,碱洗,酸洗,干燥、蒸馏,得精品。还有其它的方法吗?
[b]关于食品添加剂新品种碳酸铵、6-甲基庚醛等9种食品用香料新品种和焦亚硫酸钠等2种食品添加剂扩大使用范围的公告(2017年第1号) 根据[b]《食品安全法》[/b]规定,审评机构组织专家对食品添加剂新品种碳酸铵、6-甲基庚醛等9种食品用香料新品种和焦亚硫酸钠等2种食品添加剂扩大使用范围的安全性评估材料审查并通过。 特此公告。 附件: 1. [url=http://file2.foodmate.net/wenku/20170228w60.pdf]食品添加剂新品种碳酸铵[/url] 2.[url=http://file2.foodmate.net/wenku/20170228w60.pdf]6-甲基庚醛等9种食品用香料新品种[/url] 3. [url=http://file2.foodmate.net/wenku/20170228w60.pdf]焦亚硫酸钠等2种食品添加剂扩大使用范围[/url]备注:食品添加剂新品种:碳酸铵食品用香料新品种 :6-甲基庚醛N-(2-异丙基-5-甲基环己基)环丙基甲酰胺4-羟基-4-甲基-5-己烯酸γ-内酯糠基2-甲基-3-呋喃基二硫醚4-癸烯酸2-(4-甲基-5-噻唑基)乙醇丙酸酯4,5-辛二酮5-羟基癸酸乙酯己二酸二辛酯食品添加剂扩大使用范围:焦亚硫酸钠葡萄糖酸-δ-内酯[/b][align=right] 国家卫生计生委[/align][align=right] [/align] 2017年2月6日[b][/b]
各位同仁!请教你们一个问题,我现在在做3-甲基吡啶和3-氰基吡啶的一个液相分析,目前我把4-个标品买回来了,分别是3-甲基吡啶,4-甲基吡啶,3-氰基吡啶,4-氰基吡啶;目前的一个情况就是3-甲基吡啶和4-甲基吡啶液相无法分开,3-氰基吡啶和4-氰基吡啶无法分开,液相打出来完全重合,我用的柱子是岛津C18柱子。流动相是甲醇:异丙醇:庚烷磺酸钠溶液=7:2:91,流速:1.0ml/min,检测波长261nm,请问有谁做过这样的液相分析,能否告诉小女子一下,万分感谢!
如题,在强酸滴定强碱的实验中,所选指示剂是甲基橙,甲基橙指示终点PH=4.0左右,而甲基红指示终点PH=5.0左右。从此看出甲基红指示终点更接近PH=7.0,为什么在这个试验中,指示剂选用甲基橙,而不用甲基红呢?
RT,用正庚烷策叔丁醇和甲基叔丁基醚(MTBE)的相对校正因子。叔丁醇和甲基叔丁基醚(MTBE)的出峰时间重复性很好,但正庚烷的出峰时间飘忽不定。有时在MTBE后1分钟出来,有时候在后十几分钟出来。用的柱子是石墨化炭黑填料柱,SPAN-80减尾剂,FID.不知哪位大侠可有解,为什么会这样啊?
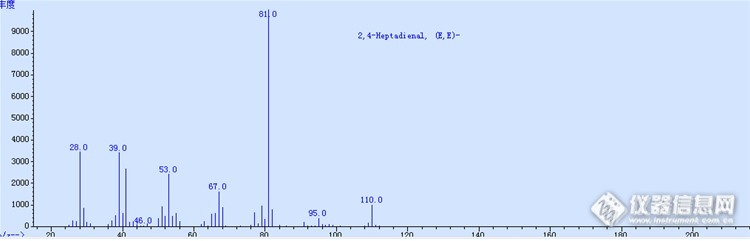
2016年前几个月都是在忙着测样,属于历史遗留问题,到现在爆发了,觉得很严重,所以样品量陡增。在检测中遇到很多问题,现在有点时间,可以慢慢和大家一起讨论讨论了。这次说说2.4-庚二烯醛的问题,样品是一个供应商的,名字就不说了。一进仪器,就发现更本不是2.4-庚二烯醛这个东西,质谱图如下:http://ng1.17img.cn/bbsfiles/images/2016/03/201603170958_587212_1060664_3.png谱库检索后发现相似度最高的为:4-Methyl-1,3-heptadiene 。而真正2.4-庚二烯醛的质谱图为:http://ng1.17img.cn/bbsfiles/images/2016/03/201603170958_587213_1060664_3.png完全不是一个东西。原料停用,同时联系供应商。供应商的答复很无语,他们早知道这个货不是我们需要的2.4-庚二烯醛,却因为错误已经造成,所以秘而不宣。现在我们发现了,他们很光棍的承认了。根据供应商提供的工艺:丙醛和巴豆醛反应生成2.4-庚二烯醛,他们认为这个未知物为:2-甲基-2.4-己二烯醛,不是谱库检索得到的那个。不管是哪个物质,总之不是我们需要的啊。这无语的供应商。当然这其中也有公司自己的责任,原料检验把关不严。现在新近的原料,都得经过GCMS分析,定性后才能入库。
流动相(甲醇:乙腈:磷酸盐)15:15:70谱图如下,如何让甲基氯异噻唑啉酮和甲基噻唑啉酮分得更开呢?[img]https://ng1.17img.cn/bbsfiles/images/2019/02/201902222125434158_9706_3489633_3.png[/img]
铺薄层板时,羧甲基纤维素钠溶液常常先过滤再与固定相混合。用多少目的筛网过滤比较合适?最近铺板,常用抽滤的方法,感觉滤后的羧甲基纤维素钠溶液似乎更稀了。常温常压下,用筛网自然过滤和用抽滤的方法过滤羧甲基纤维素钠溶液,二者有区别吗?对铺的薄层板会产生什么影响?

[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2023/11/202311151029126832_9358_5604214_3.jpg!w690x690.jpg[/img] 评估蜂蜜中羟甲基糠醛(HMF)含量的工具通常是一种重要的质量控制手段。然而,对于蜂蜜羟甲基糠醛检测仪的使用体验会受多种因素的影响。以下是一些可能影响蜂蜜HMF检测仪效用的因素: 准确性: 检测仪器的准确性是一个关键因素。仪器的设计和精度将直接影响测量结果的可靠性。高准确性的仪器通常更受欢迎,特别是在对HMF等成分进行敏感的应用中。 精确性: 仪器的精确性是指测量结果与实际值的接近程度。检测仪器的精确性对于在不同样品中重复测量非常重要。 灵敏度: HMF含量可能在蜂蜜中呈现较低浓度,因此检测仪器的灵敏度也是一个关键考虑因素。灵敏的仪器可以检测到较低浓度的HMF,对于监测质量问题和进行精细调控非常有帮助。 易用性: 检测仪器的易用性对于用户来说是一个重要因素。直观的用户界面、简单的操作流程以及易于理解的结果输出都能提高仪器的使用效率。 可靠性: 可靠性是指仪器在长期使用中的稳定性和一致性。一个可靠的检测仪器应该能够在不同条件下提供一致的结果。 成本效益: 检测仪器的成本与其性能和功能密切相关。在选择时需要权衡成本和性能,确保选择的仪器符合实际需求。 维护要求: 了解仪器的维护要求对于确保其长期性能至关重要。一些仪器可能需要更频繁或更复杂的维护,这可能会增加使用成本。 在评估蜂蜜羟甲基糠醛检测仪时,最好通过查看产品规格、用户评价、实验室或工业界的实际使用经验等途径来了解其性能。确保所选的仪器符合您的具体需求,以获得最佳的使用体验。
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url](检测器FID)做环己烷,正庚烷,TDI,MDI ,戊烷 正己烷这些,[color=#ff6666]50%二苯基-50%二苯基-50%二甲基硅氧烷共聚物毛细管柱: SH-50,rtx-50 可以吗[/color]
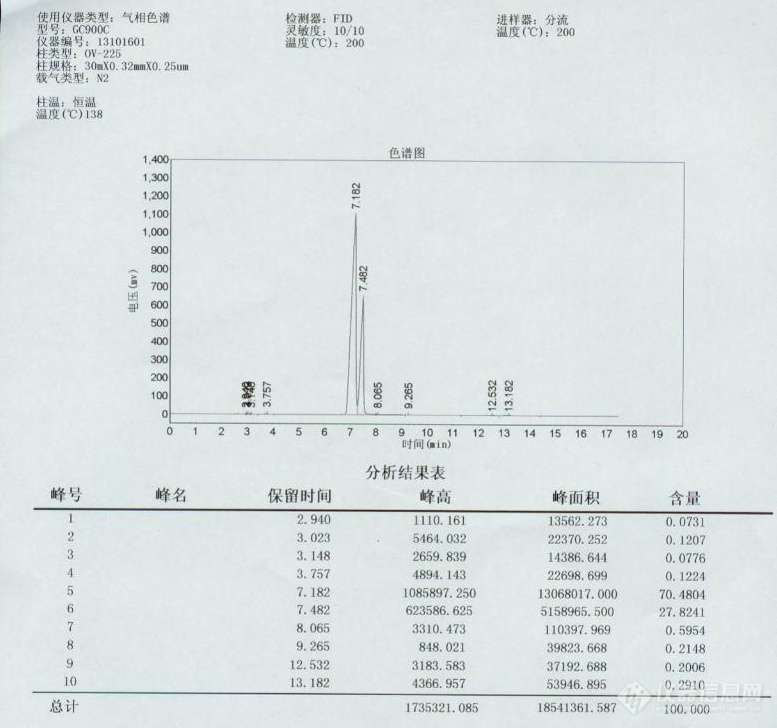
读书的时候没认真听课,所以对于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]真心不懂,请各位大侠教导教导。实验室最近新买的一台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],型号是GC9000C,柱型号是OV-225,规格:30mX0.32mmX0.25mm.来装机的人说,我们这个柱的柱温最高只能到250度。然后,我们主要是测甲基丙烯酸羟乙酯和甲基丙烯酸羟丙酯。不过我不会设温度。刚开始的时候,设的是柱温225度,检测器和进样器都是200度左右。[img=,690,557]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251136543900_1849_1853141_3.png!w690x557.jpg[/img]得出的峰都是平顶的。后来自己研究来研究去,发现只要电压上到1200以上都是平顶的。。。后来,我们的供应商提供了一张他们测的谱图给我,顺便也告诉我他们的测试条件:柱温170,检测器、进样器230.这张是供应商测的。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251136436020_7265_1853141_3.png!w690x517.jpg[/img]我按照一样的条件去测同一批羟乙酯,可是很不一样啊:首先,出峰时间是4~5分钟,电压是1200以上,平顶,含量是95左右。。。。为什么会这样?我自己用柱温138度,进样器、检测器200度测的结果是这样的:[img=,690,581]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251136256900_4608_1853141_3.png!w690x581.jpg[/img]真心搞不懂啊。。。。。然后甲基丙烯酸羟丙酯更奇怪。。。[img=,690,646]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251136104940_5072_1853141_3.png!w690x646.jpg[/img]这是什么状况啊?!!!跪求大侠大神帮忙~~~~~
各位老师新年好! 最近我在做甲基汞的实验,GB 5009.17-2003,由于我们这边没购有氯化甲基汞标准物质,只买了甲基汞(甲醇)标准溶液76μg/g浓度。用苯稀释为1ppm,上ECD检测器,没发现有目标峰。试过弱极性柱和中等极性柱了(我查的文献都基本上用这类柱子) 请问:是不是ECD这个检测器对甲基汞没有响应,要氯化甲基汞才有响应?
有谁知道对甲基苯磺酸和邻甲基苯磺酸液相色谱分离的条件么?
氮甲基苯胺、邻甲基苯胺、间甲基苯胺,对甲基苯胺在用红外光谱检测时,波长分别是多少
有时叠氮化物的亚甲基会分裂成两个一样的四重峰(化学位移略有不同)而甲基会分裂成两个一样的三重峰,为什么
水质甲基肼的测定,对二甲氨基苯甲醛分光光度法,买不到甲基肼,可以用甲基肼硫酸盐代替吗?
甲基碘磺钠盐与甲基二磺隆的沸点谢谢关心的朋友。
我现在使用的是LC-AFS测定甲基汞,因为刚接触对甲基汞的形态分析比较陌生,0价、1价、2价、3价、5价汞是代表什么,氯化甲基汞是测定甲基汞的标液吗?望各位老师不吝赐教
分析对甲基苯胺,间甲基苯胺,邻甲基苯胺,用什么毛细管色谱柱好?
有谁做过甲基汞的GC-ECD测定,标准上都是使用的氯化甲基汞(HP-5能出峰),但是发的考样又是未氯化的甲基汞,直接进样根本不出峰,不知道哪位前辈知道该如何处理。看了标准好几次,都不知道如何能让甲基汞变成氯化甲基汞…… 知道的可加QQ 382296801,希望大家多多指导……
大家好 我现在有问题想请教1-甲基萘 2-甲基萘 用什么柱子分离呀谢谢了
原水样(澄清水样,成分未知)为碱性,加入数滴甲基橙,用磷酸调pH,可在水样由碱性转变为酸性过程中,水样不显色(用pH试纸试过水样,水样已低于4.4),已确定甲基橙没有问题,其余水样均能正常显色,该水样调节PH过程中,有什么能干扰甲基橙显色?
5009.17中甲基汞的测定原理是说把甲基汞转化为无机汞和硼氢化钾反应生成氢化物后测定甲基汞的含量。方法里要配置两种形态的汞混合标液,为什么要配两种形态的汞混合标液?
现在的柱子3甲基吡啶和四甲基吡啶分不开,大家好你们给推荐一种分离较好的柱子吧!谢谢!
六甲基二硅胺烷和三甲基氯硅烷作为硅甲基化试剂与醇的反应机理! 另外,六甲基二硅胺烷可以与醇反应,为什么还要加入三甲基氯硅烷?三甲基氯硅烷不是也可以与醇反应吗?请指教!
次甲基蓝和亚甲基蓝是不是一种东西?问了几个人答案不一致,大家讨论一下!听说过次氯酸 亚氯酸,应该是有区别的!大家的意见呢?
用气相色谱检测奶粉中的肌醇时,用的是肌醇与三甲基氯硅烷 六甲基二硅胺烷 NN二甲基酰胺等硅烷化试剂,我想请教一下大神们肌醇与这些试剂的反应产物是什么,能提供相关的反应原理或者反应式吗??
用液相原子荧光检测甲基汞时,甲基汞与二价汞分不好,请教大家都用的哪家的柱子什么规格的?流动相比例?还有一个问题是L-半胱氨酸要求是生化试剂,不知道在哪里能买到,用优级醇代替可以吗?请路过的高手指点
在看书时发现有亚甲基兰和次甲基兰,不知道是否是一种物质,还请各位指教!







 我要推广仪器
我要推广仪器
 下载APP
下载APP