瑞士万通的905自动电位滴定仪,搭配光度电极。用EDTA滴定镍钴锰溶液的总量。如果滴定溶液中不加盐酸羟胺,初始电位很低很低,突越很不明显,加入盐酸羟胺初始电位就有700多mv,正常。但是观察溶液颜色,加入盐酸羟胺前后基本无变化。这是什么原因?滴定用的是可见光波长。
[em0715] 请教大家一个问题查资料说,盐酸羟胺与羟胺的一些化学性质相似,可以采用盐酸羟胺来代替后者吗?怎样才能在实验室制备自由羟胺,有篇文献说采用醇钠中和,但是没有具体的操作步骤和用量,不知哪位大侠能否提供一个具体的操作规程多谢大家了[em0703]
请问专家,羟胺和盐酸羟胺有什么区别,若有区别,有什么办法可使盐酸羟胺变为羟胺,请各位不吝赐教,衷心感谢!!!
我是搞水处理的,请高手们指教液相中的羟胺和联胺怎么测呀?
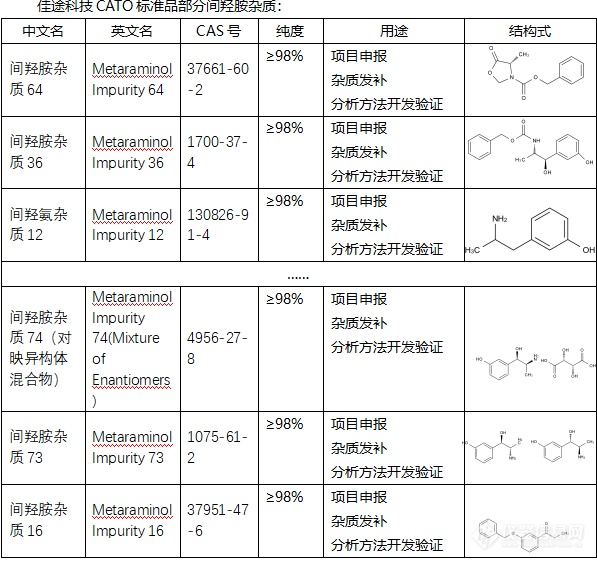
间羟胺是一种有机化合物,主要用作还原剂和抗氧化剂,也用于一些特殊的有机合成反应。然而,它也是某些药物和化妆品制造过程中的杂质,如果含量过高,可能对人体健康产生不利影响。间羟胺杂质如果未经妥善处理,可能会导致药物或化妆品的质量下降,甚至影响药效。一些间羟胺化合物对人体有毒,长期或大量接触可能会引起皮肤和眼睛的刺激,甚至影响肝脏和肾脏,或引发神经系统和呼吸道问题。除此之外,间羟胺对环境也有潜在危害,过量的间羟胺可能污染水源,对水生生物和环境产生破坏。CATO标准品格外关注间羟胺杂质的存在是十分重要的,需要通过科学严谨的检测和控制技术,保证药物和化妆品的质量和安全。[img=,597,562]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052113508399_5763_6381668_3.png!w597x562.jpg[/img]
最近做了1批水样,水中可能有铁离子和铬离子。按照水质监测方法说的,加盐酸羟胺可以有效去除干扰。我想问下 盐酸羟胺是应该时候加入去除干扰,是在消解完水样后加入还是在加完1+9盐酸后加入才对?(PS:能简单解释下盐酸羟胺反应原理吗- - )
HPLC法测定羟甲烟胺的含量 来源: 作者:武玉敏,李大伟 ,李云霞 摘要:目的:采用高效液相色谱法测定羟甲烟胺的含量。方法:以C18化学键合硅胶柱分离羟甲烟胺, 以甲醇-磷酸二氢钠(pH=6.0)(10:90)为流动相,UV检测波长261nm 进行测定。结果:平均回收率为99.76%,RSD为0.29%(n=5)。羟甲烟胺进样量与吸收峰面积呈良好的线性关系。线性范围0.1~0.6mg/ml。结论:该方法测定羟甲烟胺的含量,结果准确,重现性好。关键词:HPLC;羟甲烟胺;含量测定羟甲烟胺为利胆保肝药,并具有抑菌作用,临床上广泛用于胆囊炎、胆管炎,亦可用于病毒性肝炎的辅助治疗。国家药品标准中用碘量法测定其含量。本文试用高效液相色谱法测定羟甲烟胺的含量,取得满意的效果。1 仪器与试药岛津LC-10AD高效液相色谱仪,岛津SPD-1OAV紫外检测器,色谱工作站:天津奥特赛恩斯。羟甲烟胺对照品(自制,含量为99.8%),羟甲烟胺为山东山大康诺制药有限公司生产(批号040508 040510 040512),甲醇为色谱纯,磷酸二氢钠为分析纯。,2 实验方法2.1色谱条件色谱柱:BDS-C18(150mm×4.6mm,5μm);流动相:甲醇-磷酸二氢钠(pH=6.0)(10:90);流速:1.0ml/min;检测波长:261nm;柱温:30℃。2.2 溶液配制2.2.1对照品溶液:取105℃干燥2h的羟甲烟胺对照品约0.1g,精密称定,置100ml容量瓶中,加甲醇适量,振摇使溶解,加甲醇稀释至刻度,摇匀。超声5min,用甲醇定容,作为对照品贮备液。精密量取对照品贮备液10ml至50ml量瓶中用甲醇稀释至刻度。2.2.2供试品溶液:取研细的本品约1g,精密称定,置25ml量瓶中,加适量甲醇溶解并稀释至刻度,超声处理5min,放冷,用甲醇定容,过滤,弃去初滤液,取续滤液。即得。2.3系统适用性试验分别取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图。羟甲烟胺峰保留时同约为6.5min,理论塔板数以羟甲烟胺峰计算为4300。3 实验结果 3.1线性关系试验 精密量取对照品贮备液(1mg/ml)1.0、2.0、3.0、4.0、5.0、6.Om1分别置lOml容量瓶中,加甲醇稀释至刻度,摇匀。每个浓度点进样两次测定,各进样10μl,以峰面积为纵坐标,浓度为横坐标进行线性回归,得回归方程为:Y=0.08041x一0.01456 r=0.9999,线性范围为0.1~0.6mg/ml。3.2精密度考察 依次取同一对照品溶液(0.2mg/ml)连续进样5次,每次10μl,峰面积积分值得RSD=0.24%。3.3重复性试验 取同一羟甲烟胺样品5份,按供试品液制备方法制备,依法测定。结果羟甲烟胺含量RSD为0.37%,表明本方法重现性好。3.4稳定性试验 取同一样品溶液(批号040508),在0、1、4、12、24、48、72h分别进样10μl,依法测定,结果7次测定羟甲烟胺含量RSD为0.32%,表明样品溶液在3d内稳定。3.5回收率试验 取羟甲烟胺对照品适量,按样品测定方法测定含量,计算回收率。3.6样品测定 取浓度为0.2mg/ml的样品溶液用0.45μm微孔滤膜滤过,各进样lOμl,读取峰面积值,按外标法计算含量。4 讨论羟甲烟胺的流动相溶液置UV-2450分光光度计上,在200~400nm波长范围内扫描,结果在261nm的波长处有最大吸收,故本法的检测波长选用261nm。本实验采用高效液相色谱法测定羟甲烟胺的含量,方法操作简便,结果准确,重现性好,灵敏度高,可作为实际生产中质量控制的方法。
不知哪位大哥有乙酰羟胺的分析方法?多谢!
我在做测定水中阴离子表面活性剂,用的方法中有一个试剂是 盐酸羟胺-铁试剂:取20g盐酸羟胺,加入60mgFe2+,溶于200ml水中,这里的Fe2+是什么啊,我用的七水和硫酸亚铁行吗!!谢谢,谢谢!

空气中己内酰胺的测定方法 羟胺-氯化铁比色法 羟胺-氯化铁比色法1 原理已内酰胺和碱性羟胺反应生成异羟肟酸,再与三价铁反应形成红棕色络合物,比色定量。2 仪器2.1 冲击式吸收管。2.2 抽气机。2.3 流量计,0~5L/min。2.4 具塞比色管,10ml。2.5 水浴锅。2.6 分光光度计。3 试剂3.1 硫酸羟胺溶液,C〔(NH2OH)2H2SO4〕=2mol/L。3.2 氢氧化钠溶液,C(NaOH)=5mol/L。3.3 盐酸溶液,C(HCl)=2.5mol/L。3.4 盐酸溶液,C(HCl)=0.1mol/L。3.5 氯化铁溶液,200g/L。取20g氯化铁(FeCl36H2O)溶于盐酸溶液(3.4),并稀释至100ml。有混浊时过滤。3.6 标准溶液:称取0.5000g己内酰胺,用水溶解稀释至100ml,此标准储备液1ml含5mg己内酰胺,冰箱可保存1个月。临用前取5ml储备液,用水稀释至50ml,配成1ml含500?g己内酰胺标准溶液。4 采样串联两个各内装5ml水的冲击式吸收管,以3L/min的速度抽取15L空气。5 分析步骤5.1 对照试验:同采样,将吸收管装好吸收液带至现场,但不抽取空气,照样品分析。5.2 样品处理:用吸收管中的吸收液洗涤进气管内壁三次,取1.5ml溶液于10ml具塞比色管中供测定。5.3 标准曲线的绘制:取7只10ml具塞比色管,按表1配制标准管。表1 己内酰胺标准管的配制[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201417_52375_1625938_3.jpg[/img]5.4 测定:向样品管和标准管各加2ml硫酸羟胺溶液(3.1)和0.5ml氢氧化钠溶液(3.2),摇匀,加塞。于沸水浴中加热75min。冷却后加1ml盐酸溶液(3.3),摇匀,再加入1ml氯化铁溶液(3.5),振摇1min后于波长500nm下比色测量吸光度。以标准管己内酰胺含量对吸光度(扣除空白吸光度)作图,绘制标准曲线。样品的吸光度减去空白后,由标准曲线查出己内酰胺含量。6 计算X=5(C1+C2)/1.5V0式中:X——空气中己内酰胺浓度,mg/m3;C1、C2——分别为第1,第2吸收管所取样品中己内酰胺的含量,微克;V0——标准状况下的样品体积,L。7 说明7.1 本法的检测限为20微克/1.5ml。当己内酰胺浓度分别为25、100和500微克时,变异系数分别为1.7%、0.8%和1.3%。合并变异系数为1.23%。采样5min,取1.5ml样品分析,其测定范围为4.5~110mg/m3。7.2 如空气中己内酰胺仅有蒸气和烟状态时,可用多孔玻璃板吸收管采样,流量为1L/min。7.3 铁络合物颜色稳定性较差,显色后20min内比色完毕。如果样品较多可分批加氯化铁溶液(3.5)。加氯化铁后应充分强烈振摇1min以便把气泡全部赶走。
关于盐酸羟胺高效液相检测方法:我现在的条件下盐酸羟胺峰受到溶剂峰的干扰,而这个溶剂峰在空白溶液中是没有的.目前的条件乙腈含量为5%,请教大家如何改善盐酸羟胺和溶剂峰的分离.
加入盐酸羟胺与不加盐酸羟胺的值有区别么?
铬黑T指示剂的配制过程中,盐酸羟胺的作用是什么
各位我想请教以一下盐酸羟胺和高锰酸钾的反应方程式是如何进行的?????
[align=left] 意想不到盐酸羟胺的不稳定性对实验结果的影响[/align][align=center] [/align][align=left]在前面的二篇中提及了Tris的检测,经过近一个月的努力,顺利完成了Tris的方法开发和验证。今天谈谈另一个化合物的检测验证,盐酸羟胺,在Tris快完成之际,接到另一家客户的要求,检测某一药物中残留的盐酸羟胺。[/align][align=left]天下哪有这么好的事,盐酸羟胺和Tris结构类似,理论上检测方法可以完全套用,除了淋洗液和柱子不一样外,后面的补液模式可以完全照抄!分离方法可以基本按照Tris 的方式(阳离子分离,柱后补碱,安培检测)。[/align][align=left]在开始做之前,我问了对方,盐酸羟胺的稳定性,稳定。同时,我上网简单查了一下,没有提及不稳定的事,这样完全照搬Tris的检测模式,OK。[/align][align=left]在对方厂家测试人员的协助下,按照设计的方案,排好序列,连续进样80针就够了,时间大约14-16小时,为了加快速度,通宵进行,由于担心仪器晚上会突然断开(ICS5000)我通宵查看系统,果然半夜里软件连接突然断了一次,重新连接后就没再断开了,在连续进样的过程中,我感觉有点不对,很低浓度下,怎么没有盐酸羟胺的峰(理论计算是有呀,能看到),峰感觉在变小,似乎不稳定,半夜里只能继续做下去了。[/align][align=left]第二天在分析大批数据后,终于感觉到不对了,盐酸羟胺会分解!所有实验结果泡汤了!赶快上网查资料,的确有明确提及盐酸羟胺不稳定,但如何稳定没说呀!查到相关分析的一篇类似文献,提及用流动相溶解,但其数据和结果似乎表明其检测限比我们高近10倍,他们做的很有问题。怎么办?样品不稳定这样实验做起来就麻烦了,羟胺,盐酸羟胺,我明白了,没有单独的羟胺的实际样品,只有盐酸羟胺,这就意味着只要盐酸的存在,不稳定的羟胺变成了稳定的盐酸羟胺,酸性条件稳定,也理解了论文用流动相溶解的意思。[/align][align=left]重新设计实验,盐酸羟胺稀释不用水稀释而是用跟淋洗液几乎一样的淋洗液浓度进行稀释,在酸性条件下,这样可以避免盐酸羟胺的分解。重新配溶液上机,在仅仅改变样品的溶解稀释方式,整个实验的重复性非常好,盐酸羟胺峰面积稳定回收率合格,而实验仅仅改变了一点!![/align][align=left]在实验中,由于做检测限,盐酸羟胺的浓度非常低,羟胺水解的效应非常明显,当浓度高时,不是很明显,这就是为什么一开始做了半天没发觉的缘故。对比这二个实验,Tris和盐酸羟胺结构类似,测试方法几乎一致,但完全照抄在实际中却遇到问题,说简单很简单,说复杂也不简单。Tris作为缓冲液肯定是稳定的,这是二者的差异。[/align][align=left]这也验证了做方法学的主要关键之一,样品的稳定性必须先验证。[/align]
测定化妆品中重金属元素,汞和镉需要在消解液中加入盐酸羟胺,而砷和铅的消解液中不需要加盐酸羟胺,加盐酸羟胺的目的是什么?为什么砷和铅不需要加?如果汞和镉不加盐酸羟胺会出现什么样的情况?盐酸羟胺加与不加,对最后的结果有没有影响?
[size=16px]请教下大佬们,[color=#3333ff]硫化物会和联氨/羟胺发生反应吗[/color]?联氨(N2H4,-2价),羟胺(NH2OH,-1价),硫化物(-2价,最低价态)虽然从氧化还原价态上,联氨和羟胺不会被硫化物还原为更低价的NO3-(+5价)或NO2-(+3价),但它会不会被硫化物氧化为NH4+(-3价)呢?能不能查到相应的化学反应方程式?求助!!谢谢大家!![/size]
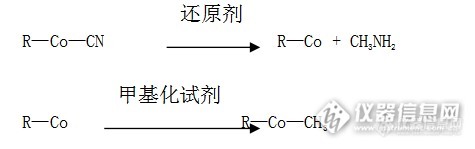
[align=center][b]甲钴胺精制过程的研究[/b][/align][align=left][b]摘要:目的[/b]甲钴胺作用与维生素B12(氰钴胺)类似,能够维持人体血细胞的正常形态与功能,并维持神经纤维功能的完整,促进轴索内轴流和轴索再生及髓鞘形成,因此对神经轴突传递延迟和神经传递物质的减少有很好的恢复作用。开展甲钴胺原料药工艺技术研究开发与生产,生产出成本低、质量高、对环境污染小的产品,满足临床用药需求,具有重大现实意义。[b]方法[/b]选用了甲钴胺专用精制介质及优化的生产工艺条件,[b]结果[/b]本课题选取了甲钴胺专用的精制介质,该介质对于甲钴胺精制具有较强的专属性,能够有效降低甲钴胺的有关物质,能够制备高纯度的甲钴胺。极大提高了临床用药的安全性。优化了大生产的工艺条件,提高了产品品质,降低了成本。[b]结论[/b]生产出的产品质量好、收率高,甲钴胺收率平均约93%,生产过程中污染物产生量小,拥有很高的实用价值。[/align][b]关键词:[/b]甲钴胺原料药;生产工艺;精制纯化 在氰钴胺的水溶液中,用还原剂进行还原,脱去CN[sup]-[/sup]生成VB[sub]12[/sub]的还原态,其中CN[sup]-[/sup]以甲氨(CH[sub]3[/sub]NH[sub]2[/sub])的形式脱去。还原态的VB[sub]12[/sub]带有一个负电荷,用亲核的甲基化试剂进行甲基化反应,生成甲钴胺,反应式如下:[align=center][img=,425,133]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152018_02_1626619_3.png[/img][/align] 式中:R—Co—CN为氰钴VB[sub]12[/sub],R—Co为VB[sub]12[/sub]还原态,R—Co—CH[sub]3[/sub]为甲钴胺国内多数药厂在甲钴胺生产工艺上都处于技术的一个更新阶段,生产厂家的生产质量参差不齐,多数较大生产厂商都能够达到药典的质量要求,但是对于小的药厂来讲,甲钴胺生产工艺还需要进一步改进。本研究将针对上述各种不足对大生产关键工艺条件进行优化。最终使得甲钴胺的合成工艺既高效,副产物又少,达到质量与收率都提高,同时又不产生固废与异味,减少环保压力。[b]1 甲钴胺精制介质的选择[/b] 现代医药技术要求原料药的色普纯度较高[sup][[/sup][sup]1[/sup][sup]][/sup],对于维生素B12品种来说国际上一般要求其相关物质以高效液相色谱法检测不得大于2.0%。我们为了使得甲钴胺原料药在国际上更具有竞争力,将目标定为相关物质控制在1.0%以内。因此选取一种能够有效去除甲钴胺的相关物质的精制介质是我们这个课题中的重要内容。利用目前已有的精制介质,有50%的甲钴胺产品的有关物质不能达到这个要求。为此我们重点研究了甲钴胺的理化性质,根据它的特征基团的极性常数以及常见相关杂质的特征基团的极性常数,我们提出在一些常用的精制介质的单体中加入某些化学基团,利用甲钴胺和它的相关物质在这种特别的精制介质上的分配系数的不同,从而达到分离的目的。为此课题组与介质生产厂家合作生产了5种精制介质:JZ-1、JZ-2、JZ-3、JZ-4、JZ-5。对5种介质的精制性能进行了试验研究。试验方法如下: 在5个相同的精制柱中,分别加入等量的5种精制介质,取等体积的同一批次甲钴胺溶液,以相同流速通过5个精制柱,分别用等体积的展层剂以相同流速展层,再用等体积的解析剂以相同的流速解析,获得结晶原液,用高效液相色谱法测定其相关物质及甲钴胺的收率。试验情况见表2-1。[align=center]表1-1 精制介质的分离效果[/align][align=center][img=,650,176]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152015_01_1626619_3.png[/img][/align] 注:表中分离效果是指在精制介质上杂质色带与主色带的分离程度。 从表1-1可以看出,精制介质ZJ-3分离相关物质的能力最好。为进一步考察精制介质ZJ-3的分离能力,采用3批甲钴胺做验证试验,其结果见表1-2。[align=center]表1-2 精制介质分离效果表[/align][align=center][img=,660,129]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152018_01_1626619_3.png[/img][/align] 从表1-2中数据可以看出:精制介质JZ-3对甲钴胺分离效果好、收率高(平均收率在96%以上)、可操作性强,因此,选取JZ-3作为该工艺的精制介质。 (1)专用精制介质的特性 目前国际上的VB12提取与精制行业普遍采用的层析介质是XAD1180大孔型树脂和三氧化二铝,这两种介质对所有的VB12品种均有效,但是其选择性不好,分离效果差。为了验证我们创制的专用精制介质良好的分离性能,我们做了对比试验。试验方法:用相同的甲钴胺料液,分别在专用精制介质、XAD1180树脂、三氧化二铝上进行展层精制,考察它们对相关物质去除的程度和收率。具体数据见表1-3。[align=center]表1-3 专用精制介质与其它介质的效果对比数据[/align][align=center][img=,690,113]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152022_01_1626619_3.png[/img][/align] 从上表数据可以看出,我们创制的甲钴胺专用精制介质的性能大大优于国际上普遍采用的分离介质。 (2)小试工艺的验证试验 选用同一批氰钴胺作为原料,分别用原生产工艺与本课题小试确定的工艺同时进行投料反应,重复进行三次,对本课题确定的工艺进行验证。试验数据见表1-4。[align=center]表1-4 验证试验数据 [/align][align=center][img=,650,231]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152023_01_1626619_3.png[/img][/align] 从表1-4可以看出:小试在同一批原料进行的重复试验中,产品的有关物质去除率均比原生产工艺有较大提升,产品质量均达到并超过多国药典的要求,平均收率达到93%以上,超过了日本专利中报道的87%的水平及现有工艺90%的水平。说明生产工艺的稳定性高,可操作性强。[b][b][b][b]2 工艺参数的优化[/b][/b][b][b]2.1高纯氮气的通气量的确定[/b][/b][/b][/b] 据文献报道,反应液中氧的浓度大于0.1ppm时,反应液中的氧会将还原态的VB[sub]12[/sub]氧化为羟钴胺。反应式如下:[align=center][img=,458,39]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152025_01_1626619_3.png[/img][/align] 因此,必须控制反应时中氧的浓度。本研究采用向反应液中通入保护性气体,以脱除反应液中的氧,避免氧化反应的发生。试验方法:反应液中的氧主要来自于溶解原料的水,所以以水代料进行通气试验。对于通气的实际操作我们发现对于容积只有1m3的反应器,在搅拌状态下通入高纯氮气的最大流速只能固定在2000L/min左右,过大会使反应液逃液,为此我们以高纯氮气的流速和通入时间为考察对象,测定反应液氧气含量。因此,选择10组通气量,分别通入与反应液同体积的水中,在5、10、15、20、25、30、40、60 min 时测定水中氧的浓度。试验数据见表3-1。试验数据见表2-1:[align=center]表2-1 一定通气量时不同时间氧的浓度[/align][align=center][img=,650,463]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152028_01_1626619_3.png[/img][/align] 从表2-1数据可以看出,通气时间在20分钟以内能够使水中的氧浓度低于0.1ppm的通气量为:大于600L/min 。将600、800、1000、1200 L/min的通气量作为进一步试验的通气量,以考察能使反应液中氧浓度达到0.1ppm以下的时间对甲钴胺转化率及羟钴胺含量的影响(在高效液相图谱中羟钴胺的百分比越高就说明反应液中氧气浓度越高)。试验数据见表2-2。[align=center]表2-2 不同通气量对转化率的影响[/align][align=center][img=,650,161]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152030_01_1626619_3.png[/img][/align] 从表3-2数据可以看出甲钴胺的转化率并不能说明这个问题,这是因为生成的羟钴胺含量太小,而测定精度不够造成该数据不能有效表征这个问题;而从图谱中可以明显看到通气量与羟钴胺在图谱中的含量差异。根据羟钴胺的含量可知600L/min、800L/min的通气量比较合适,而通气量为1200L/min时,在通气时发生冲料现象,致使反应无法进行。通气量为1000L/min时,产生大量的泡沫,因此该研究选取的通气量为600~800L/min,且通气时间定为30min比较合理。[b][b]2.2搅拌形式的选取[/b][/b] 在该反应中,搅拌效果会在一定程度上影响甲钴胺的转化率。在相同搅拌速度下,我们以浆式、窝轮式及折叶式搅拌形式进行试验,考察搅拌方式对转化率的影响。试验数据见表2-3。[align=center]表2-3 搅拌形式对甲钴胺转化率的影响[/align][align=center][img=,650,304]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152031_01_1626619_3.png[/img][/align] 从表2-3数据可以看出,选用折叶式搅拌形式较合适。[b][b]2.3精制介质展层速度的确定[/b][/b] 由于生产中使用的柱子高度在3米以上,塔板高度与理论板数不能与小柱子同日而语,所以展层速度对于生产展层工艺来说比较重要。其展层效果直接影响甲钴胺的色谱纯度与含量的高低。对于确定的精制介质,若展层剂比例一定,柱子填料一定,装量高度一定的情况下,则其展层速度对分离效果的影响较大,它们遵循范弟姆特动力学方程[align=center]H=A+B/U+C×U[/align][align=left] 其中:H为塔板高度 A为涡流扩展项 B为分子扩散项 C为传质阻力系数 U为展层速度[/align] 如果U过小,可认为C项不起作用,可忽略,这样因B/U值大而导致H值过大,分离效果降低;如果U过大,可认为B可忽略,因C×U 值增大,而导致H值过大,分离效果亦降低。为得到好的分离效果,对介质JZ-3的展层速度进行了试验研究,试验方法为:在相同的精制柱中,加入适量的的精制介质JZ-3,取等体积的反应液,吸附于这个精制柱中,分别用同一比例的丙酮水溶液以不同的10组流速进行展层,而后用同一解析剂以相同的流速解析,用高效液相色谱法测定结晶原液的相关物质及甲钴胺的收率。试验数据见表2-4。[align=center]表2-4 展层速度对甲钴胺有关物质的分离能力[/align][align=center][img=,600,333]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152037_01_1626619_3.png[/img][/align] 由表2-4中数据可以看出:展层速度为700 L/ h时有关物质最低,此时收率为93.8%,而展层速度为600 L/ h和800L/ h时其有关物质与收率也比较与之接近。因此将600~800L/ h的速度作为该工艺的展层速度。采用以上试验确定的工艺参数,设计并建设了甲钴胺生产性试验装置。稳定生产后。连续生产10批的数据见表2-5。从表2-5的数据可以看出:使用相关物质在2.3%以上的氰钴胺作原料生产的甲钴胺,相关物质全部在1.0%以下,收率平均达到93%以上,甲钴胺含量达到98.5%以上。[align=center]表2-5 生产结果统计[/align][align=center][img=,650,296]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152039_01_1626619_3.png[/img][/align] 从表2-5大生产的批次统计结果可以看出,小试确定的试剂及工艺条件,在大生产中可以生产出稳定的高品质的甲钴胺产品,同时又可以做到清洁生产,降低环保压力的目的。[b]3 质量研究[b]3.1 各国药典标准及产品内控标准[sup][[/sup][sup]3][/sup][/b][/b][align=center] 表3-1 国内市场标准(CP)[/align][align=center][img=,650,317]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152045_01_1626619_3.png[/img][/align][align=center]表3-2 日本市场标准(JP)[/align][align=center][img=,650,273]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152046_01_1626619_3.png[/img][/align][align=center]表3-3美国市场标准(USP)[/align][align=center][img=,650,336]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152055_02_1626619_3.png[/img][/align][b][b]3.2新工艺开发前后产品主要质量指标对比[/b][/b][align=center]表3-4 工艺开发前后产品结果对比[/align][align=center][img=,650,216]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152057_01_1626619_3.png[/img][/align] 以新工艺制备的三批样品的HPLC图谱及色谱纯度结果见图3-1,图3-2,图3-3。[align=center][img=,650,468]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152059_01_1626619_3.png[/img] [/align][align=center]图3-1 甲钴胺样品-1HPLC图谱[/align][align=center][img=,619,471]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152101_01_1626619_3.png[/img][/align][align=center]图3-2甲钴胺样品-2HPLC图谱[/align][align=center][img=,650,456]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152101_02_1626619_3.png[/img] [/align][align=center]图3-3甲钴胺样品-3HPLC图谱[/align] 综上结果,本课题开发工艺所生产的产品质量在有关物质项明显优于开发前工艺。[b][b]3.3 稳定性试验[/b][/b] 为进一步确定开发后工艺所生产产品的质量稳定性情况,按照相关法律法规要求,将前三批大生产产品列入了长期36个月及加速6个月稳定性考察研究。参照已上市产品的储运条件为10-30℃,参照相关指导原则在温度25±2℃,相对湿度60±5% 的条件下进行,避光保存。以第0个月,3个月、6个月、9个月、12个月、18个月、24个月、36个月为考察时间点,进行重点项目检测。[b][b]3.4 稳定性考察结果统计与分析[/b][/b] 比较重点考察项目,包括性状、含量、有关物质、水分等相关指标不同考察时间节点的变化是否存在显著性差异,其差异的变化范围应在药典质量控制范围之内。[align=center]表3-5甲钴胺加速试验结果[/align][align=center][img=,650,422]http://ng1.17img.cn/bbsfiles/images/2017/09/201709152102_01_1626619_3.png[/img][/align] 表3-5结果表明,按照开发的新工艺生产的产品在重要质量指标:性状、含量、有关物质、水分都符合相关药典要求。目前该项研究仍在进行中。在已完成的加速稳定性考察过程中,产品重点质量指标均符合各相关药典规定;[b][b]4 结果与讨论[/b][/b] (1) 本课题选取了甲钴胺专用的精制介质,该介质对于甲钴胺精制具有较强的专属性,能够有效降低甲钴胺的有关物质,能够制备高纯度的甲钴胺。极大提高了临床用药的安全性。 (2) 优化大生产工艺条件,提高产品质量,降低成本。 总之,以本课题研发的生产工艺生产的甲钴胺原料药具有收率高、质量好、成本低、清洁污染少等优点。在满足患者临床用药安全性、提高公司市场竞争力方面具有重大意义。[b]参考文献[/b]AKAIKEA, TAMURAY,SATOY, YOKOTAT. Protective Effectsofa Vitamin B12 Analog, Methylcobalamin,Against Glutamate Cytotoxicity in Cultured lortical Neurons. EurJPharmacol, 1993,241(1):1-6.石山忠似等, 高纯度B12的制备方法 Chemistry and Biochemistry of B12. Published simultaneously in Canada. Edited by RUMA BANERJEE ,Department of Biochemitry, University of Nebraska Lincoln,NE. 367-379. 国家食品药品监督管理局.[color=#333333] 《[/color]已上市化学药品变更研究的技术指导原则(一)[color=#333333]》[/color],国食药监注242号:7-15.

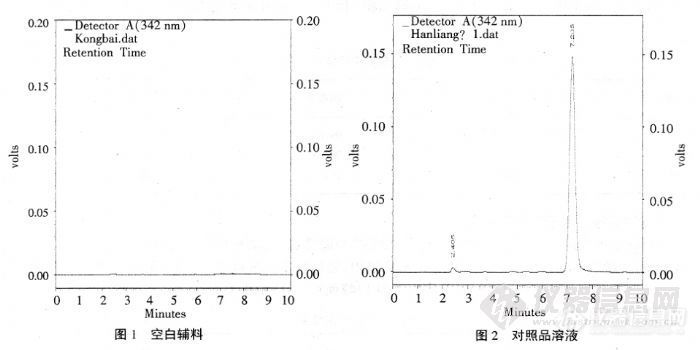
问题:2015中国药典要求甲钴胺峰与羟钴胺峰的分离度是?答案:药典要求甲钴胺峰与羟钴胺峰(相对保留时间约为0. 2)的分离度应大于20。【活动奖励】幸运奖(2钻石币):千层峰(注册ID:jxyan)sixingxing(注册ID:v2889187)ZHAOGUANGXI(注册ID:ZHAOGUANGXI)http://ng1.17img.cn/bbsfiles/images/2015/11/201511201512_574505_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511201513_574506_1610895_3.jpg积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================甲钴胺样品制备 制备方法含量测定系统适用性溶液:甲钴胺对照品(0.5 mg/mL),水溶解,自然光下放置10 min,即得。对照品溶液:取甲钴胺对照品适量,精密称定,加流动相溶解并定量稀释制成每1 mL中约含50 μg的溶液,即得。分析条件色谱柱Inspire C18 150 x 4.6 mm,5 μm (Cat#:81001)流动相0.03mol/L 磷酸二氢钾溶液(用0.2 mol/L 氢氧化钠溶液或磷酸调节pH 值至4.5):乙腈=84:16流速1.6 mL/min柱温30 ℃检测器UV 342 nm进样量20 μL色谱图含量测定对照品溶液http://ng1.17img.cn/bbsfiles/images/2015/11/201511201000_574453_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 分离度 1 12.461 50544 2107 6070.328 0.972 -- 系统适用性溶液 http://ng1.17img.cn/bbsfiles/images/2015/11/201511201001_574454_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度* 1 1.158 880662 152634 712.423 1.086 -- 2 12.197 65167 2812 6186.804 0.975 27.812 *药典要求甲钴胺峰与羟钴胺峰(相对保留时间约为0. 2)的分离度应大于20,甲钴胺峰与相邻杂质峰之间的分离度应符合要求。本品种同时使用了DiamonsilC18、DiamonsilC18(2)两款色谱柱,在药典规定条件下进行检测,均满足药典要求。
我们现在建立同时分析甲醇,乙醇和二甲基羟胺的方法,采用的是热电公司的气相色谱仪,柱子是毛细管柱,分析甲醇和乙醇的效果非常满意,但是二甲基羟胺的效果不好,精密度不高,请问是不是我们的柱子不适合分析二甲基羟胺呢?期待高人的解答啊!
硫酸羟胺的含量怎样测?
有人做过工作场所钒的N-肉桂酰-邻-甲苯羟胺光度法吗?求标准曲线
标题:5.1 甲钴胺注射液含量的方法学研究作者:常 明 1, 李 晶 1, 武玉洁 2, 张文双 2( 1.石家庄学院 化工学院, 河北 石家庄 050035; 2.石家庄栢奇制药有限公司, 河北 石家庄 050035)摘要: 摘 要: 以乙腈- 甲醇- 0.05 mol /L磷酸二氢钾溶液( 10∶ 20∶ 70) (用磷酸调节 pH 值为 4.0)为流动相, 采用迪马钻石 C18( 250 mm× 4.6 mm, 5 μ m)色谱柱及紫外检测器, 建立了甲钴胺注射液中甲钴胺含量的反相高效液相色谱检测方法. 柱温: 40℃; 检测波长为 264 nm; 流速: 1.0 mL /min. 本方法测定的日内精密度为 0.59%, 日间精密度为 0.67%, 回收率为 99.7%~ 100.5%, 在 160~ 240 μ g /mL范围内线性关系满足要求( r=0.999 9) ; 结果表明方法准确、 操作简单、 专属性强, 可用于定量测定甲钴胺注射液含量.http://ng1.17img.cn/bbsfiles/images/2012/07/201207161648_377914_2379123_3.jpg
请问关于总氮的测定(碱性过硫酸钾消解法).如果有六价铬或者是三价铁的影响,就往水样中加入1-2mL的盐酸羟铵溶液,用来去除干扰,盐酸羟胺消解前加,还是消解后加?消解前加空白也加的话 空白会增加
盐酸羟胺溶液保存是否要避光?即是否要用棕色试剂瓶保存?
我用盐酸羟胺代替氯化亚锡作还原剂,但是不生成络合物也不显色。请大侠赐教显色条件。
我测量的主要是自来水,在工程师给我的方法中测量汞时不需要加溴酸钾-溴化钾溶液和盐酸羟胺溶液,但是GB5750-2006里是需要加的,起到消解转换作用。这里请问各位大佬:1、我们测量的主要是自来水,溴酸钾-溴化钾溶液和盐酸羟胺溶液是否需要加?如果不需要的话,那在什么条件下才需要加?2、玻璃器皿浸泡液是10%硝酸还是30%硝酸?
[color=#444444]今天做实验,是处理样品然后做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],每个样加10毫克盐酸羟胺我加入的量差的比较多觉得,不知道对实验有没有影响呀。[/color]
盐酸羟胺的饱和乙醇溶液怎么配置呢?
有人做过工作场所钒的N-肉桂酰-邻-甲苯羟胺光度法吗?求标准曲线







 我要推广仪器
我要推广仪器
 下载APP
下载APP